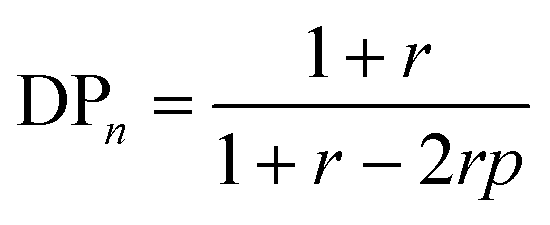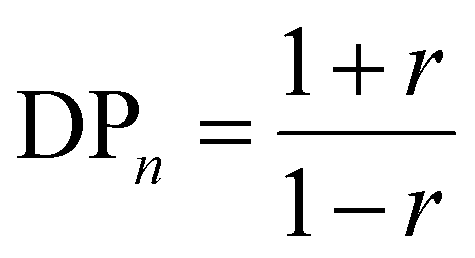DOI:
10.1039/D4PY01091D
(Review Article)
Polym. Chem., 2025,
16, 11-26
Recent progress in nonstoichiometric step-growth polymerization
Received
30th September 2024
, Accepted 2nd December 2024
First published on 3rd December 2024
Abstract
According to the theory established by Carothers and Flory, stoichiometric control of monomers is necessary for efficient step-growth polymerization of AA-type and BB-type monomers in a single-phase solution. This review examines recent progress in synthesizing high-molecular-weight polymers via atypical nonstoichiometric step-growth polymerization (NSSP). The reactive intermediate mechanism (RIM) and intramolecular catalyst transfer (ICT) systems are essential for efficient NSSP, generating polymers with much higher molecular weights than theoretically predicted. NSSP systems provide many advantages in fine synthetic technologies for producing complex multifunctional polymers suitable for specific applications.
 Kenta Yokawa | Kenta Yokawa received his B.Eng. degree from Yamagata University in 2023 and is currently a master's course student at Yamagata University. He has been financially supported by the Hasegawa Foundation and Nakamura-Sekizenkai Foundation. His research interests include nonstoichiometric condensation polymerization and direct arylation polymerization. |
 Tomoya Higashihara | Tomoya Higashihara received his B.Eng., M.Eng., and D.Eng. degrees from the Tokyo Institute of Technology in 2000, 2002, and 2005, respectively. He became a postdoc at the University of Massachusetts Lowell in 2005 and was promoted to Assistant Professor at the Tokyo Institute of Technology in 2008. During 2010–2013, he joined the PRESTO program supported by JST. He was then promoted to Associate Professor and Professor at Yamagata University in 2013 and 2019, respectively. His research interests include condensation polymerization and organic electronics. He has contributed to publishing 270 papers, 21 books, and 39 patents. |
1. Introduction
According to the theory established by Carothers and Flory, the relationship between the number-average degree of polymerization (DPn), the stoichiometric ratio between A–X–A and B–Y–B monomers (r), where A and B are reactive functional groups and X and Y are monomer repeating units, and the extent of the reaction (p) can be defined as eqn (1) in step-growth polymerization of A–X–A and B–Y–B monomers.1–3 Indeed, eqn (1) accurately predicts DPn when the polymerization is performed in a single-phase solution.| | 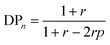 | (1) |
When p = 1 at the end of polymerization, eqn (1) can be summarized by eqn (2).
| | 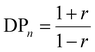 | (2) |
r should be close to 1 to obtain high-molecular-weight polymers based on eqn (2), which requires strict stoichiometry of the feed ratio between A–X–A and B–Y–B. This theory assumes that the reactivity of the functional groups in the monomers and oligomers remains unchanged during polymerization. However, high-molecular-weight polymers can be obtained even under nonstoichiometric conditions if the reactivity changes during polymerization.
A deep understanding of atypical nonstoichiometric step-growth polymerization (NSSP) has been motivated not only by academic interests but also by technical advantages for creating functional polymer materials as follows:
(a) The strict balancing of feed ratios of A–X–A and B–Y–B is unnecessary, possibly enabling facile synthesis of high-molecular-weight polymers without specific conditions.
(b) Even under stoichiometric feed ratios of A–X–A and B–Y–B, it is sometimes difficult to maintain the actual stoichiometry of their functionalities during polymerization, especially under conditions such as side reactions, volatile monomers, and precipitation of generating polymers. NSSP has the potential to overcome these difficulties.
(c) The polymerization time can be shortened by increasing the concentration of one of the monomers. When employing the A–X–A monomer with low reactivity, excess loading of the A–X–A monomer toward B–Y–B can be a viable option for increasing the reaction rate between A–X–A and B–Y–B.
(d) There is potential to introduce the same functional group at the chain ends of the resulting polymers such as A–polymer–A or B–polymer–B, which can be utilized as starting polymers for synthesizing, e.g., well-defined triblock or multiblock copolymers. Notably, conventional step-growth polymerization generally results in a mixture of functionalities at the chain ends, A–polymer–A, A–polymer–B, and B–polymer–B.
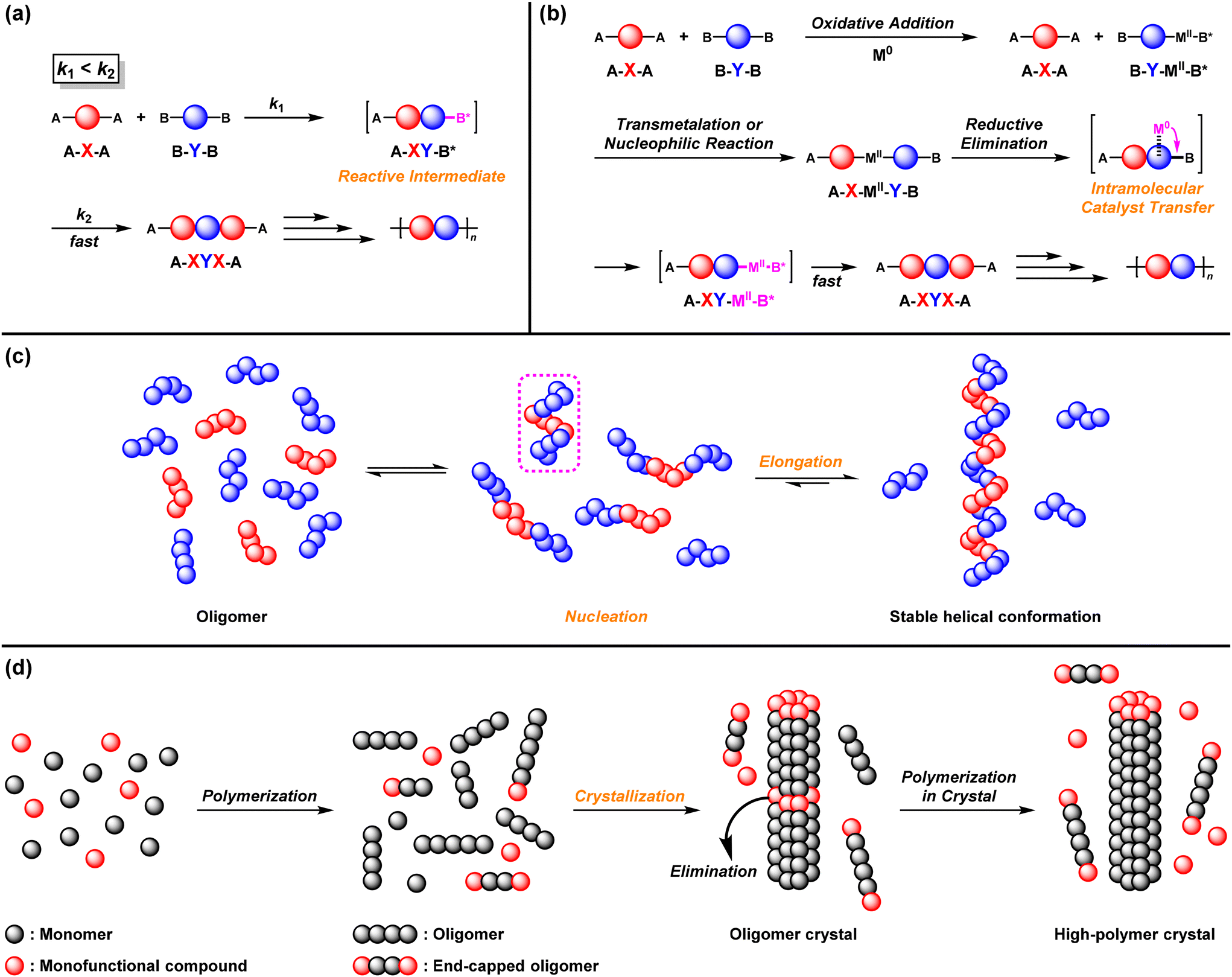
A typical strategy for achieving NSSP using A–X–A and excess B–Y–B monomers is based on a reactive intermediate mechanism (RIM) (Fig. 1a).4–39 The B group of the intermediate A–X–Y–B* dimer becomes more reactive than that of the original B–Y–B after the first reaction between the A–X–A and B–Y–B monomers. This accelerates the second reaction between the A–X–Y–B* dimer and A–X–A to afford the A–X–Y–X–A trimer in the presence of excess B–Y–B. Because both ends of the polymer intermediates are always capped by less-loaded A, polymerization proceeds even under the imbalanced stoichiometry of A–X–A and B–Y–B (excess).
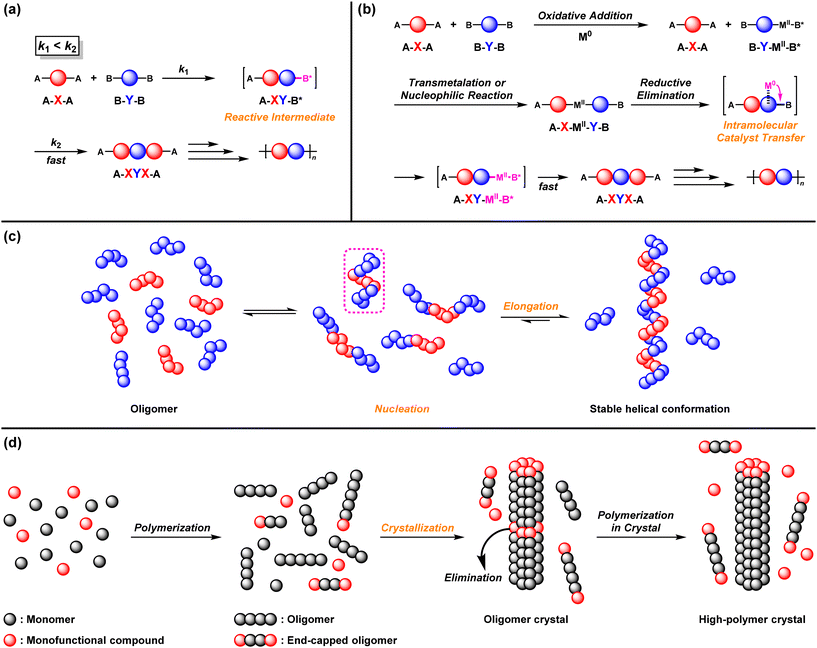 |
| | Fig. 1 Strategies for NSSP: (a) reactive intermediate mechanism (RIM), (b) intramolecular catalyst transfer (ICT), (c) folding-driven nucleation–elongation mechanism,68 and (d) reaction-induced crystallization mechanism.76 | |
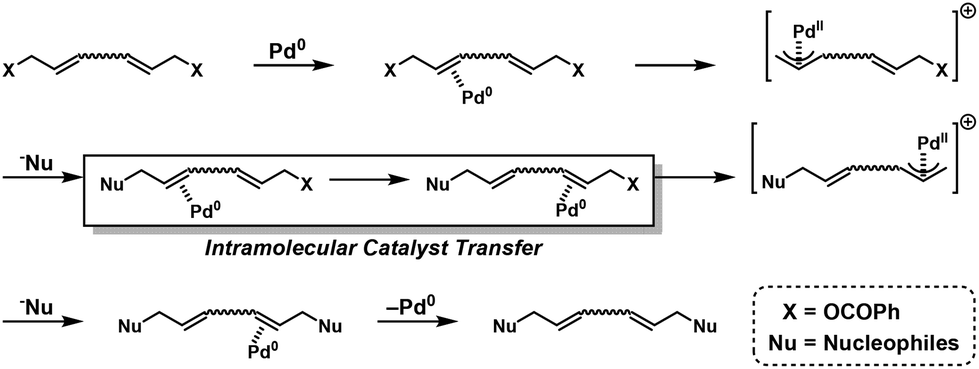
Intramolecular catalyst transfer (ICT) is another strategy for achieving NSSP (Fig. 1b).46–67 In the ICT system, B–Y–B is first activated via oxidative addition (OA) of a zerovalent metal catalyst (M0) to afford intermediate B*–MII–Y–B, which reacts with A–X–A via transmetalation (TM) or nucleophilic reaction (NR) to afford intermediate A–X–MII–Y–B. Subsequently, MII intramolecularly transfers to the next of the B group to afford the activated A–X–Y–MII–B*. Then, A–X–Y–MII–B* readily reacts with A–X–A in the presence of excess B–Y–B, followed by reductive elimination (RE) to afford A–X–Y–X–A, with regeneration of M0.
A folding-driven nucleation–elongation mechanism has also been reported to realize NSSP (Fig. 1c). Indeed, imine metathesis polymerization under an imbalanced monomer feed ratio of A–X–A and B–Y–B was developed.68,69 In this system, when the generated oligomers reach a specific size, they adopt a stable helical conformation in solution (nucleation), and the polymer starts to grow (elongation) while maintaining its helical structure. Since excess monomers cannot participate in the growth of intermediate polymers possessing a specific shape, the resulting polymers with similar DPn values are obtained regardless of which monomer is used in excess.
A reaction-induced crystallization mechanism has been reported to achieve NSSP (Fig. 1d).70–75 High-molecular-weight aromatic polyesters and polyimides were prepared under nonstoichiometric conditions in the condensation polymerization of A–X–A and B–Y–B, which involved a change in polymerization phases. In this system, oligomers are formed by (addition)–condensation reactions in solution, causing oligomer crystals to precipitate. The polymerization proceeds via transesterification or transmetalation reactions within the oligomer crystals, in which chain end groups are excluded from the inner solid phase and moved to the outer liquid phase. Polymerization is not terminated via transesterification or transmetalation reactions, affording high-molecular-weight polymers, even if the oligomer chain ends are capped with excess monomers.
Although comprehensive reviews of NSSP based on the systems above have previously been published,76–82 this review mainly focuses on the recent progress in NSSP systems for synthesizing high-molecular-weight polymers. It includes two sections of RIM and ICT systems to achieve NSSP.
2. Reactive intermediate mechanism
As described in the Introduction, NSSP using A–X–A and excess B–Y–B monomers can typically be achieved using RIM.4–39 The initial reaction between A–X–A and B–Y–B produces the reactive intermediate A–X–Y–B*, with a reaction rate constant of k1 (Fig. 1(a)). The second reaction of A–X–Y–B* with a deficit of A–X–A proceeds with a much higher rate constant of k2 (k1 ≪ k2).
2.1. Nucleophilic aromatic substitution reaction
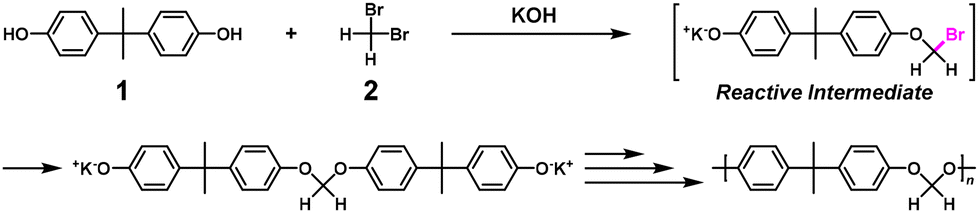
Nonstoichiometric condensation polymerization (NSCP) using bisphenol A (1) and excess methylene dibromide (2) was reported to obtain high-molecular-weight aromatic polyformals in the presence of a base and phase transfer catalyst (Scheme 1).4,5 The geminal dibromo substituents are a key chemical structure leading to the high ratio of reaction rate constants, κ (= k2/k1).
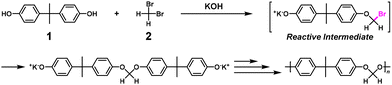 |
| | Scheme 1 Representative synthesis of polyformal via NSCP using 1 and excess 2 based on RIM.5 | |
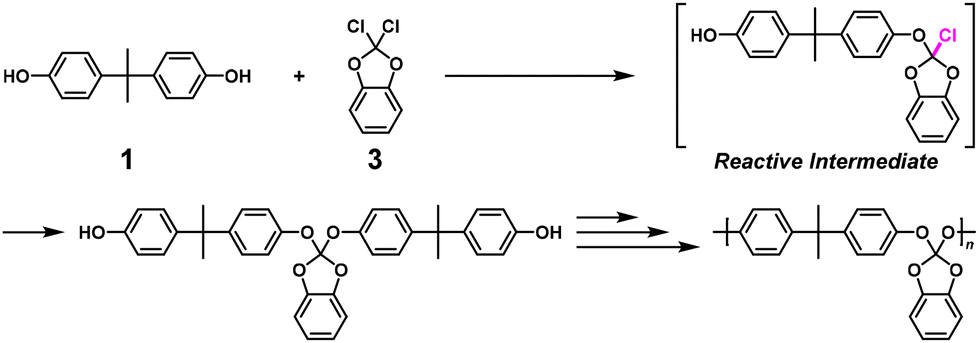
High-molecular-weight polyorthocarbonate via NSCP using 1 and a 1.67-fold excess of 2,2-dichloro-1,3-benzodioxole (3) was also synthesized with a number-average molecular weight (Mn) of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, which was much higher than the calculated value based on eqn (2) (Mn = 693) (Scheme 2).6 Notably, 3 possesses geminal dichloro substituents, which was important for obtaining a high κ value of 27 in a model reaction.
000, which was much higher than the calculated value based on eqn (2) (Mn = 693) (Scheme 2).6 Notably, 3 possesses geminal dichloro substituents, which was important for obtaining a high κ value of 27 in a model reaction.
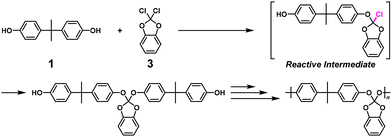 |
| | Scheme 2 Synthesis of polyorthocarbonate via NSCP using 1 and excess 3 based on RIM.6 | |
Alternatively, polythioether was successfully synthesized via NSCP using 4,4′-thiobis(benzenethiol) (4) and excess 2 (Scheme 3).7
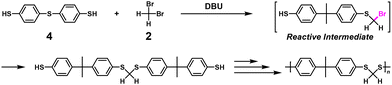 |
| | Scheme 3 Synthesis of polythioether via NSCP using 4 and excess 2 based on RIM.7 | |
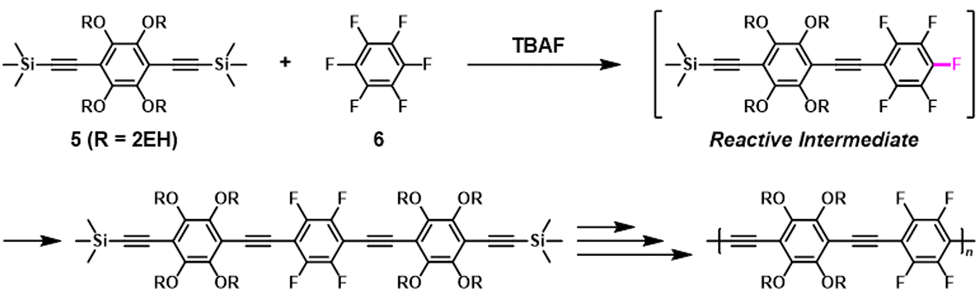
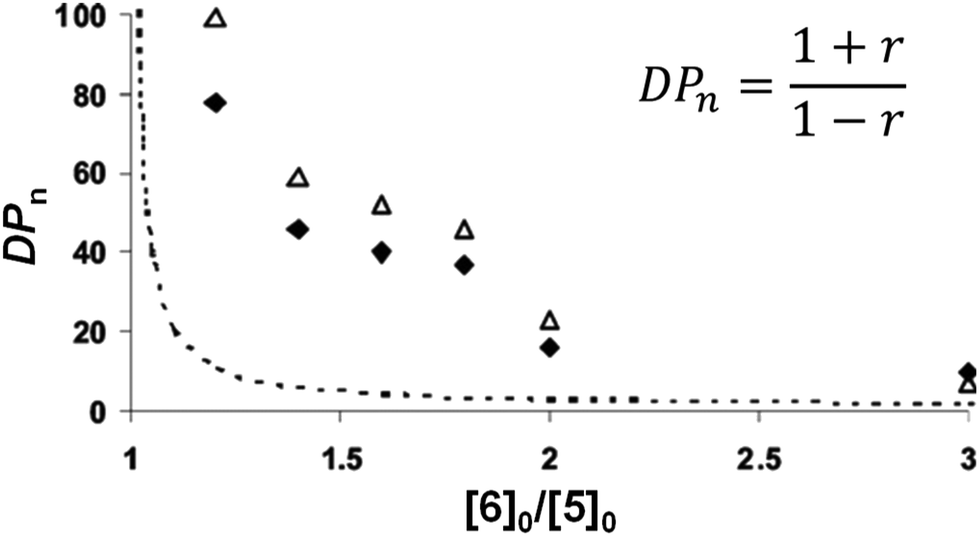
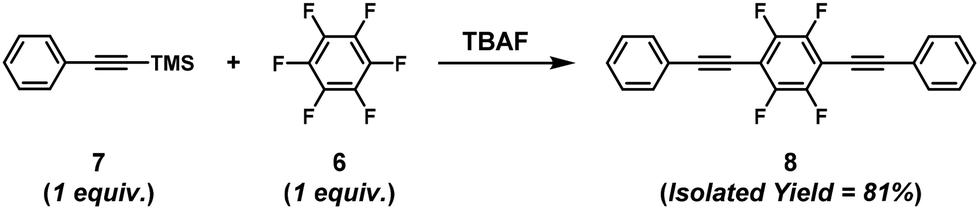
The synthesis of π-conjugated poly(phenylene ethynylene)s with alternating aryl–perfluoroaryl units was achieved via condensation polymerization of 1,2,4,5-tetrakis(2-ethylhexyloxy)-3,6-bis(2-(trimethylsilyl)ethynyl)benzene (5) and hexafluorobenzene (6) in the presence of a catalytic amount of fluoride ions (Scheme 4).39 Using an excess of 6 toward 5 afforded poly(phenylene ethynylene) with a much higher DPn value than expected based on the Carothers/Flory theory (Fig. 2). The model reaction between trimethyl(phenylethynyl)silane (7) and 5 ([5]0/[7]0 = 1/1, a 2-fold excess of reactive fluorine elements in 5 toward silyl groups in 7) afforded a diadduct compound 8 in an 80% yield (Scheme 5). This result indicates that the second substitution reaction is faster than the first, supporting the RIM of the proposed polymerization system. To the best of our knowledge, this is the first report on the one-pot synthesis of π-conjugated polymers via NSCP based on RIM.
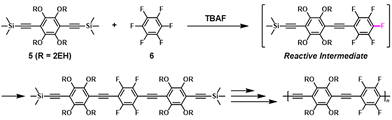 |
| | Scheme 4 Transition-metal-free synthesis of π-conjugated poly(phenylene ethynylene)s via NSCP between 5 and 6.39 | |
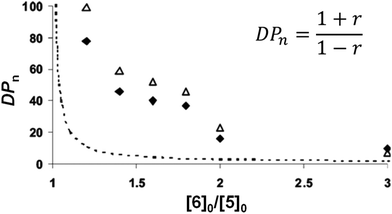 |
| | Fig. 2 DPnvs. molar ratio [6]0/[5]0, estimated using nuclear magnetic resonance end-group analysis (◆) and size exclusion chromatography (△), compared to the Carothers/Flory theory (dashed line). Reprinted after modification with permission from ref. 39. Copyright 2008 American Chemical Society. | |
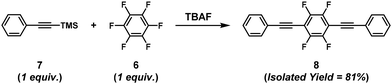 |
| | Scheme 5 The model reaction between 7 and 6.39 | |
2.2. Electrophilic aromatic substitution reaction
NSCP based on superacid-catalyzed polyhydroxyalkylation reactions was reported.8 Indeed, NSCP between excess isatin (9) and biphenyl (10) or terphenyl yielded high-molecular-weight polymers in the mixed superacid solvent of trifluoromethanesulfonic acid (TFSA) and trifluoroacetic acid. The high κ was mainly due to the high reactivity of the carbocationic intermediates, which can be controlled by tuning the acidity of the media and the basicity of the electrophilic carbonyl compounds. A polymer with ultrahigh-molecular-weight (Mn = 1140000) could be synthesized when using biphenyl and a 1.15-fold excess of 10 (Scheme 6).9 The same group reported NSCP using bisphenols and excess ketones to synthesize heteroaromatic polymers under similar conditions stated above.10
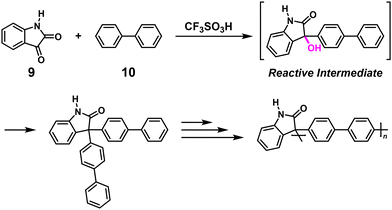 |
| | Scheme 6 Synthesis of poly(biphenyl-alt-isatin) via NSCP based on the superacid-catalyzed polyhydroxyalkylation reaction between 9 and excess 10.9 | |
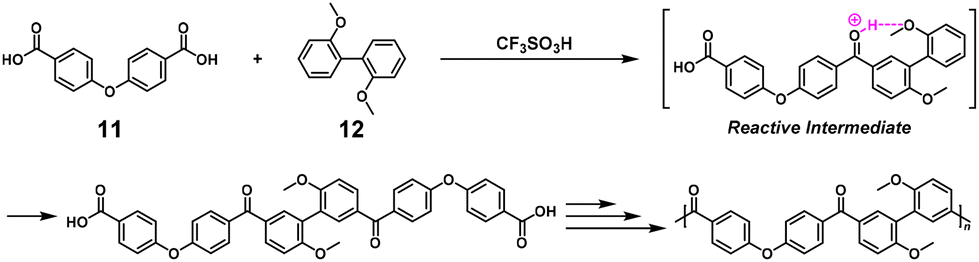
NSCP was achieved based on the Friedel–Crafts acylation reaction in TFSA for synthesizing aromatic poly(ether ketone)s (Scheme 7).11 In practice, NSCP of 4,4′-dicarboxydiphenyl ether (11) and up to a 1.5-fold excess of 2,2′-dimethoxybiphenyl (12) proceeded in TFSA to afford high-molecular-weight poly(ether ketone) (Mn = 26100–73300). The driving force for the high κ was identified as the increased reactivity of the monoacylated species caused by deprotonation in TFSA. A similar system was adopted to synthesize aromatic poly(ketone)s (Mn = 44900–94800) using aromatic dicarboxylic acid chlorides and up to a 2-fold excess of 12 in the presence of aluminum chloride.12
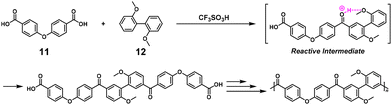 |
| | Scheme 7 Synthesis of poly(ether ketone) via NSCP based on the Friedel–Crafts acylation between 11 and excess 12 in TFSA.11 | |
2.3. Palladium-catalyzed condensation polymerization
The cascade bidirectional allylation mechanism was proposed to synthesize poly(alkylene vinylene)s via NSCP based on the Tsuji–Trost reaction between excess (Z)-but-2-ene-1,4-diyl diacetate (13) and diethyl malonate (14) (Scheme 8).15 In this report, the polymerization mechanism was explained as follows: 13 is first activated by catalytic zerovalent palladium (Pd0) via OA to obtain an intermediate allyl–PdII complex 15. 15 is then reacted with the anion of 14 (first allylation reaction) to afford an intermediate allyl–PdII complex 16via the ICT system (path A). 16 is intermolecularly attacked by the anion of 14 to form the disubstituted compound 17 (second allylation reaction), regenerating catalytic Pd0. There could be another route for the dissociation of Pd0 and the monosubstituted product 18 from their complex (path B). Because the re-oxidation reaction of Pd0 with 18 is considerably slow, a successful NSCP requires high selectivity of path A.15 For instance, poly(alkylene vinylene)s with higher DPn values of 37–66 than expected from eqn (2) (DPtheoryn = 3–21) were obtained under the nonstoichiometric condition of the monomer ratio [13]0/[14]0 = 1.1–2.0.
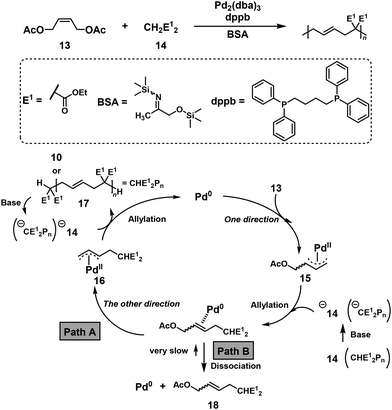 |
| | Scheme 8 Synthesis of poly(alkylene vinylene)s via NSCP between 13 and 14, and proposed polymerization mechanism.15 | |
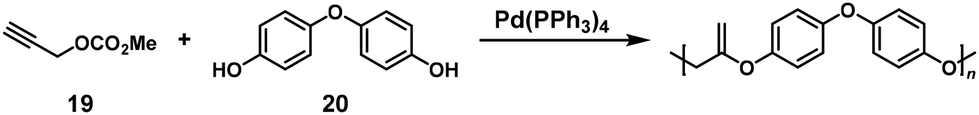
Novel Pd-catalyzed condensation polymerization of diphenol and propargyl carbonate compounds was also carried out for synthesizing polyesters with exomethylene groups. The use of methylpropargyl carbonate (19) in a 2-fold excess toward 4,4′-oxydiphenol (20) resulted in a successful NSCP system to afford relatively high-molecular-weight polyester (Mn = 3700) (Scheme 9).16 The mechanism of the NSCP system was speculated to be similar to the aforementioned report.15
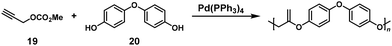 |
| | Scheme 9 Synthesis of poly(alkylene vinylene)s via NSCP between 19 and 20.16 | |
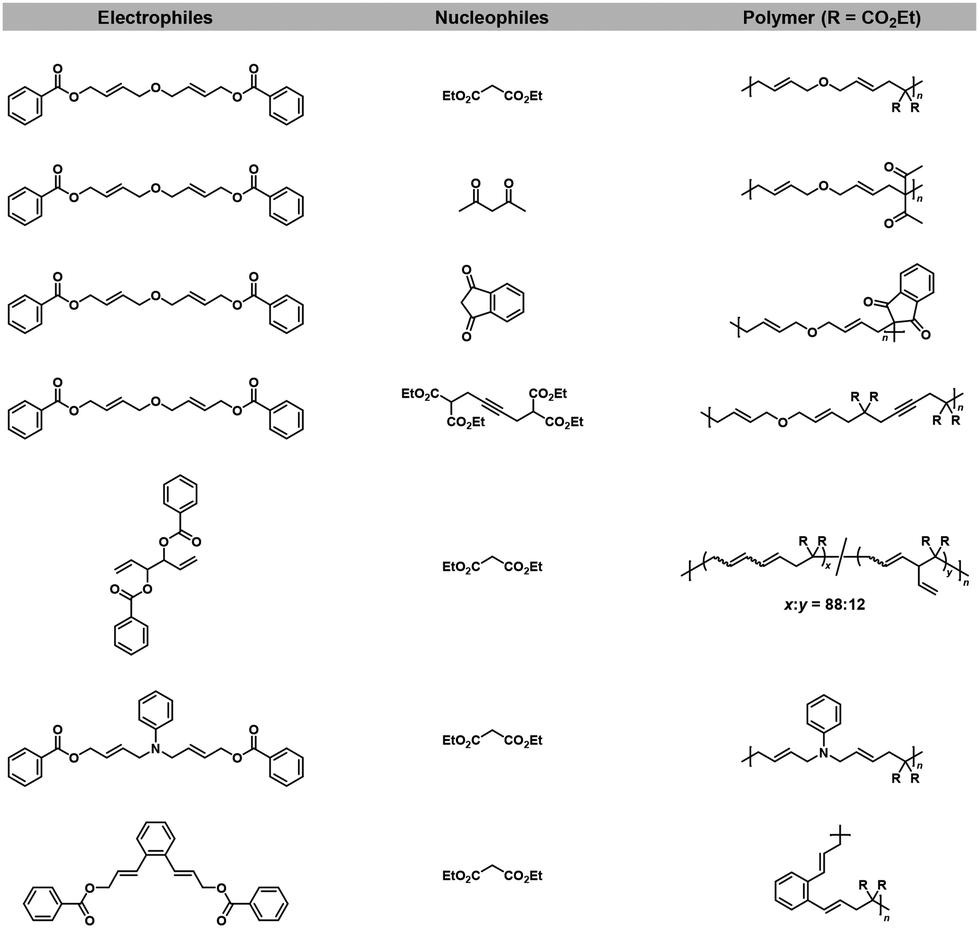
The same group also reported the synthesis of other polyethers,17,19 polyamines,18 and polyimides18via Pd-catalyzed NSCP using a wide variety of nucleophiles and excess electrophiles (Fig. 3).
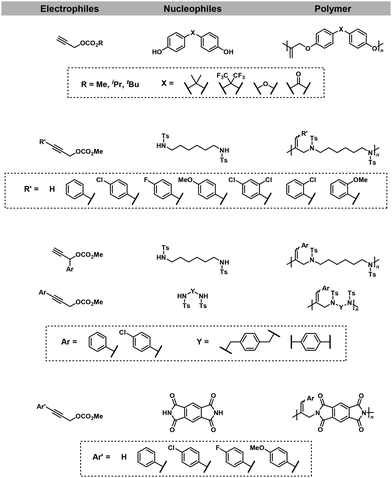 |
| | Fig. 3 Chemical structures of electrophiles and nucleophiles adaptable for NSCP based on the remote cascade double allylation strategy using the Tsuji–Trost reaction.17–19 | |
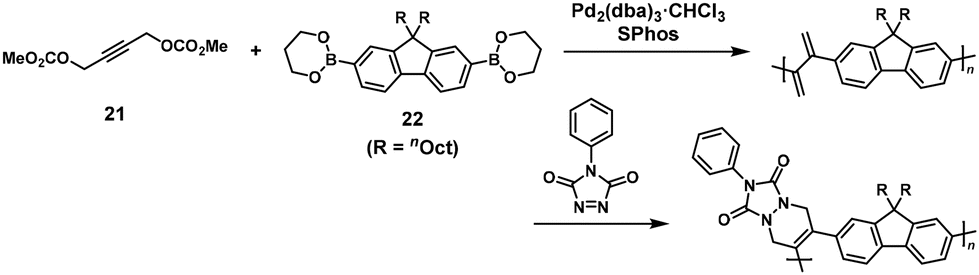
π-Conjugated polymers could also be synthesized using two-stage reactions, involving NSCP between propargylic bis(carbonate) (21) and π-conjugated monomers with diboronic acid/esters based on fluorene, carbazole, or phenylene units.20 For example, Pd-catalyzed cross-coupling polymerization of 9,9-dioctylfluorene-2,7-diboronic acid bis(1,3-propanediol ester) (22) and a 1.25-fold excess of 21 was performed in the presence of a mono chloroform complex of tris(dibenzylideneacetone)dipalladium(0) ([Pd2(dba)3]·CHCl3), 2-dicyclohexylphosphino-2,6-dimethoxybiphenyl (S-Phos), and potassium carbonate (K2CO3) to form an exomethylene-containing precursory polymer, followed by its post-functionalization via the Diels–Alder reaction with N-phenyl-1,3,4-triazoline-2,5-dione (Scheme 10). As a result, a π-conjugated polymer (Mn = 9800, Mw/Mn = 5.57) was obtained.
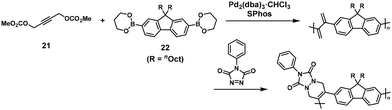 |
| | Scheme 10 Synthesis of a π-conjugated polymer via Pd-catalyzed NSCP between 22 and excess 21, followed by the Diels–Alder reaction.20 | |
2.4. Ring-closing reaction
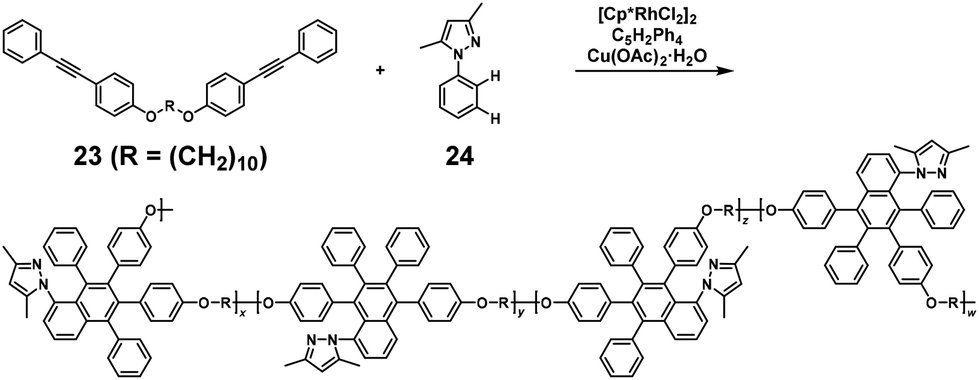
NSCP based on the rhodium (Rh)-catalyzed oxidative coupling and ring-closing reaction was performed for synthesizing polyethers. The NSCP between 4,4′-(α,ω-alkylenedioxy) bis(diphenylacetylene)s with excess phenylboronic acid or (1,1,2-triphenylvinyl)phenylboronic acid was catalyzed by a pentamethylcyclopentadienyl rhodium dichloride dimer, [Cp*RhCl2]2, and oxidants in dimethylformamide, affording polyethers with highly substituted naphthalene rings.21 A typical example is shown in Scheme 11.22 When using 1,10-bis(4-(phenylethynyl)phenoxy)decane (23) with a 2-fold excess of 3,5-dimethyl-1-phenylpyrazole (24), a high-molecular-weight polyether was obtained (weight-average molecular weight (Mw) = 35700). This successful NSCP was explained as follows: (a) coordination of 24 to the RhIIIX3 species by regioselective C–H bond cleavage of 24, affording intermediate 25 (Scheme 12), (b) insertion of 23 into the C–Rh bond of 25, forming intermediates 26 and 27 (first reaction, k1), (c) a cyclorhodation reaction at the 3′-position of the phenyl ring, producing intermediates 28 and 29, and (d) insertion of the second 23 into 28 and 29 followed by RE, affording a mixture of 23–24–23 trimers (second reaction, k2). Because intermediates 28 and 29 are more reactive than 25, k2 was much higher than k1, resulting in a high κ. Iridium-catalyzed polymerization was similarly adaptable to NSCP of 4,4′-(1,6-hexylenedioxy) bis(diphenylacetylene) and a 2-fold excess of benzoic acid or its derivatives in the presence of a pentamethylcyclopentadienyl iridium dichloride dimer, [Cp*IrCl2]2, and silver carbonate (Ag2CO3), affording high-molecular-weight polyethers (Mw up to 228700).24
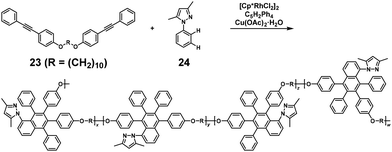 |
| | Scheme 11 Synthesis of polyethers via NSCP based on the Rh-catalyzed oxidative coupling and ring-closing reaction between 23 and excess 24.22 | |
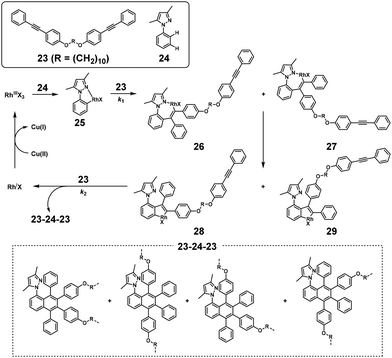 |
| | Scheme 12 Proposed mechanism for NSCP based on the Rh-catalyzed oxidative coupling and ring-closing reaction between 23 and excess 24.22 | |
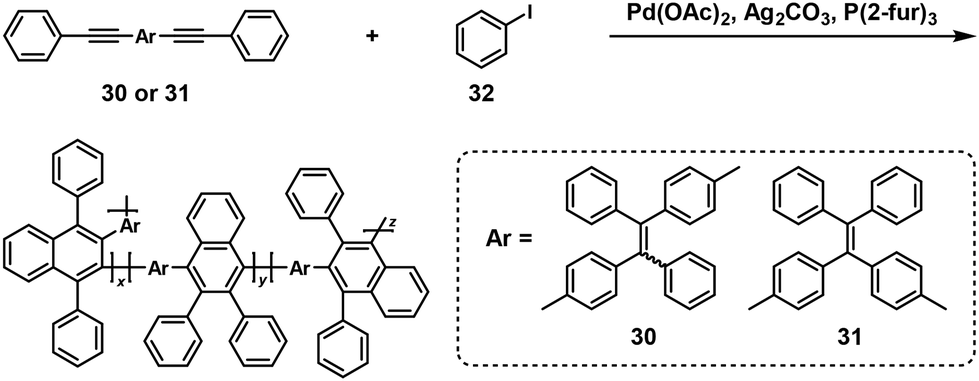
π-Conjugated polymers could also be synthesized by a similar approach.26,27 For instance, the polymerizations of internal diynes (30 or 31) and a 1.25-fold excess of iodobenzene (32) were catalyzed by palladium(II) acetate (Pd(OAc)2) and additives in o-xylene at 80 °C, affording π-conjugated polynaphthalenes with high Mw values up to 48100 (Scheme 13).26
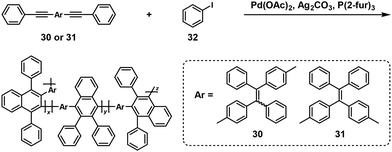 |
| | Scheme 13 Synthesis of π-conjugated polynaphthalenes via NSCP of 30 or 31 with a 1.25-fold excess of 32.26 | |
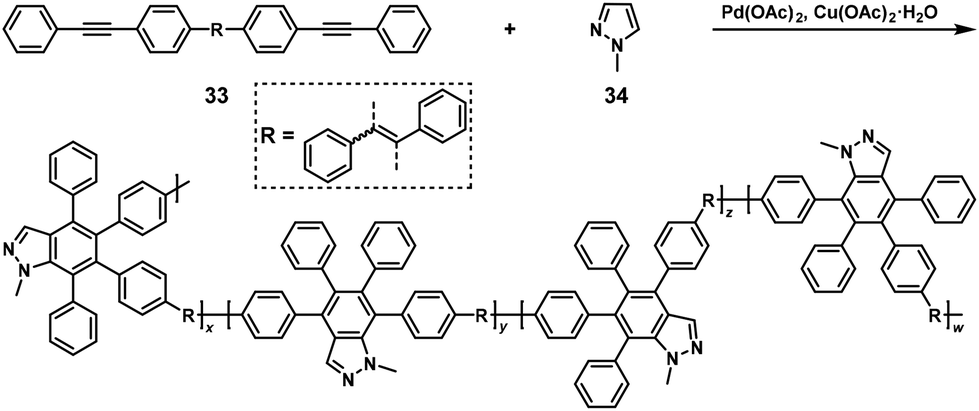
Similarly, NSCP based on the Pd-catalyzed ring-closing reaction of pyrazoles and diynes successfully produced π-conjugated polymers. Indeed, π-conjugated poly(indazole)s were synthesized via NSCP of internal diyne (33) and a 2-fold excess of 1-methyl-1H-pyrazole (34) (Scheme 14).27
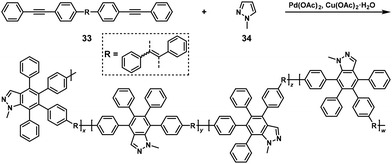 |
| | Scheme 14 Synthesis of π-conjugated poly(indazole)s via NSCP of 33 with a 2-fold excess of 34.27 | |
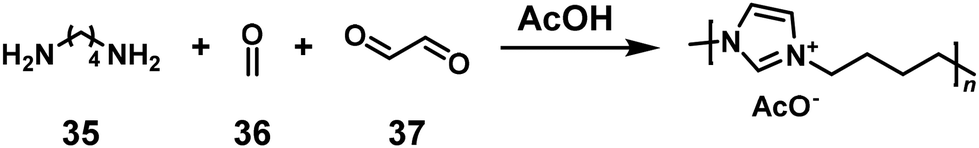
An NSCP was achieved based on another ring-closing reaction, the Radziszewski reaction, in the presence of an acid, without transition metal catalysts (Scheme 15).28 The polymerization of acetic acid (AcOH), 1,4-butanediamine (35), formaldehyde (36), and glyoxal (37) was conducted in an aqueous solution. Using excess 35 to 36 and 37 (NH2 > total C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), low-molecular-weight imidazolium-containing polymers were produced according to the Carothers/Flory theory. In contrast, when excess 36 and 37 to 35 (NH2 < total CO) was used, high-molecular-weight imidazolium-containing polymers were unexpectedly obtained (r = 0.70–0.95, Mw = 37100–44200).
O), low-molecular-weight imidazolium-containing polymers were produced according to the Carothers/Flory theory. In contrast, when excess 36 and 37 to 35 (NH2 < total CO) was used, high-molecular-weight imidazolium-containing polymers were unexpectedly obtained (r = 0.70–0.95, Mw = 37100–44200).
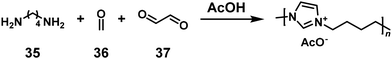 |
| | Scheme 15 Synthesis of an imidazolium-containing polymer via NSCP based on the Radziszewski reaction of 35 with excess 36 and 37.28 | |
2.5. Polyaddition reaction based on click chemistry
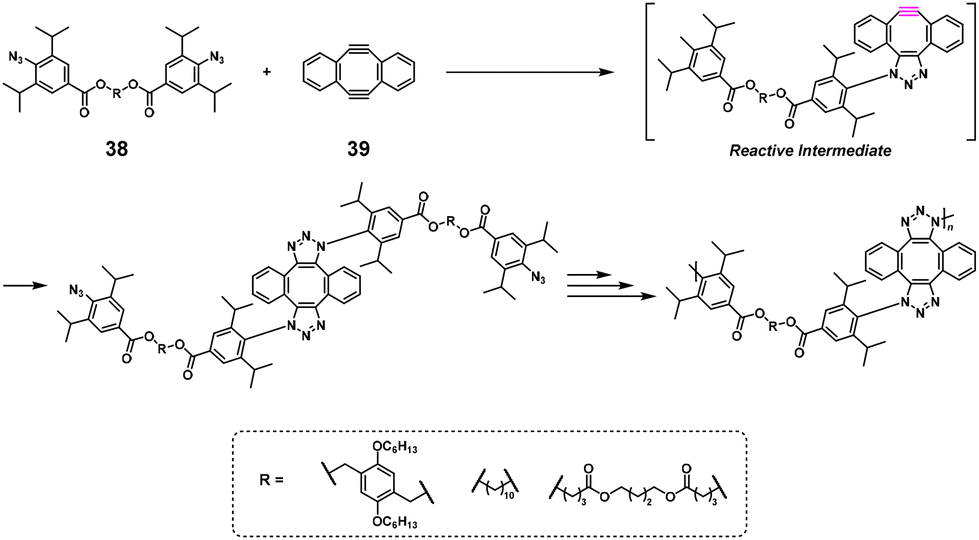
The polyaddition reaction is classified as step-growth polymerization and can be considered an environmentally friendly polymerization method because of its high atom economy. In particular, click chemistry receives much attention because of its fast and quantitative reaction efficiency. Nonstoichiometric polyaddition (NSPA) was reported based on a double-strain-promoted azide–alkyne cycloaddition reaction between a diazide compound (38) and sym-dibenzo-1,5-cyclooctadiene-3,7-diyne (39).29 In practice, high-molecular-weight 1,2,3-triazole-containing polymers (Mn = 70600–159100) were obtained using, e.g., 38 and a 1.2–2.0-fold excess of 39 (Scheme 16). The successful NSPA was explained by the activation of the second cyclization reaction after the first. Similar NSPA systems were also reported using other diazide30,31 and 1,3-dipole monomers32 with excess 39.
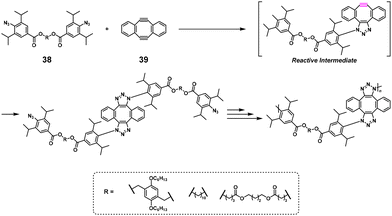 |
| | Scheme 16 Synthesis of a 1,2,3-triazole-containing polymer via NSPA based on the double-strain-promoted azide–alkyne cycloaddition reaction of 38 with excess 39.29 | |
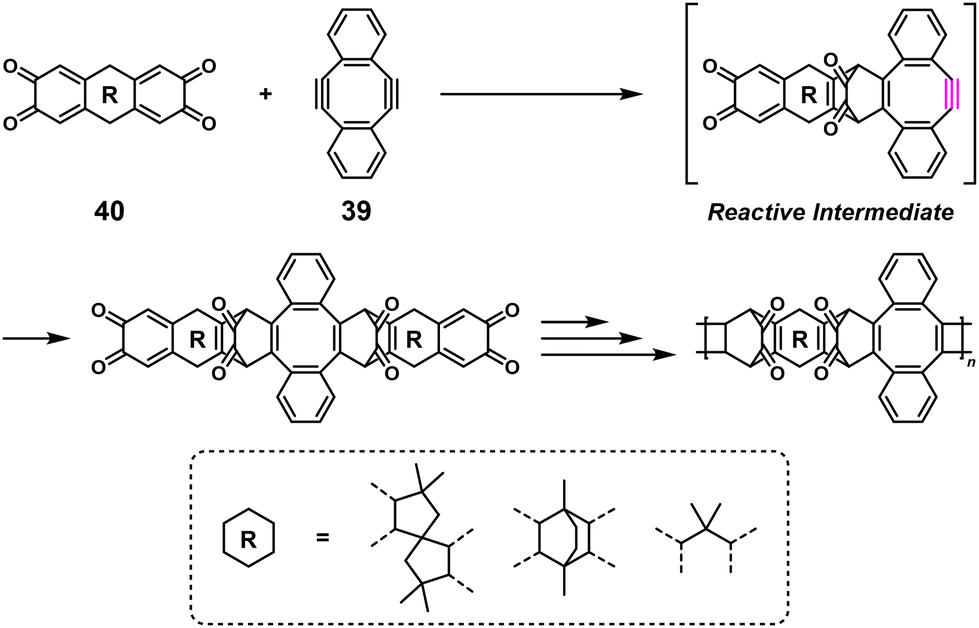
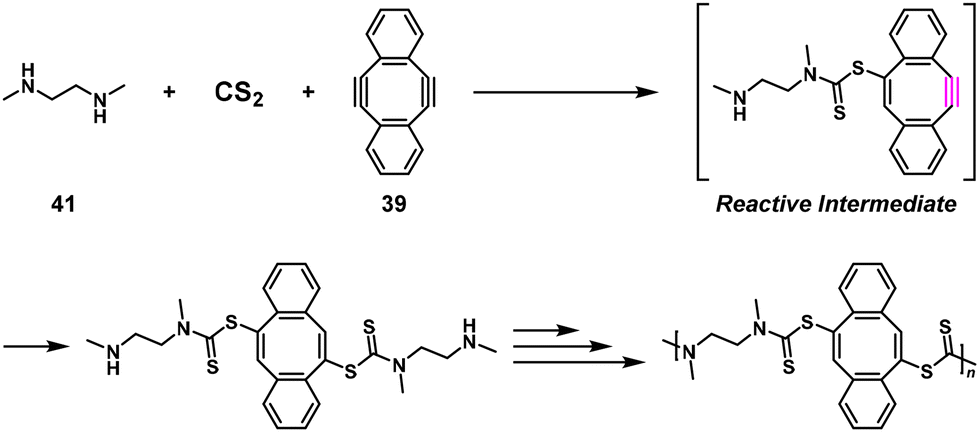
Interestingly, 39 was also adapted for NSPA based on the Diels–Alder reaction to afford high-molecular-weight ortho-quinone-containing polymers using an ortho-quinone compound (40) and a 1.1–1.5-fold excess of 39 (Scheme 17).35 The self-accelerating three-component reaction adaptable for NSPA based on the thiol–yne click reaction also afforded high-molecular-weight dithiocarbamate-containing polymers using N,N′-dimethylethylenediamine (41), carbon disulfide (CS2), and a 1.1–1.5-fold excess of 39 (Scheme 18).36
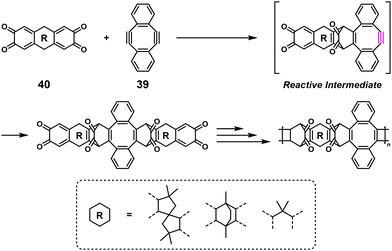 |
| | Scheme 17 Synthesis of an ortho-quinone-containing polymer via NSPA based on the Diels–Alder reaction of 40 with excess 39.35 | |
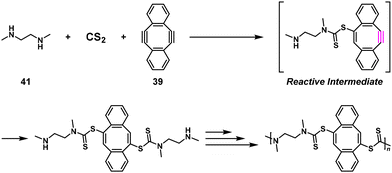 |
| | Scheme 18 Synthesis of a dithiocarbamate-containing polymer via NSPA based on the thiol–yne reaction of 41, CS2, and excess 39.36 | |
NSPA was also demonstrated based on an imine–yne click reaction.37 High-molecular-weight imidazolidine-based polymers (Mn up to 20600) were obtained using dialkyne compounds and a 1.25-fold excess of imidazoline compounds. Scheme 19 depicts the representative synthesis of an imidazolidine-based polymer employing propane-2,2-diylbis(4,1-phenylene)propionate (42) and excess 1-methyl-2-phenyl-4,5-dihydro-1H-imidazole (43).
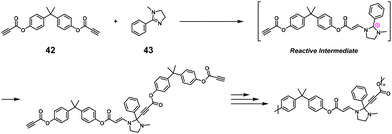 |
| | Scheme 19 Synthesis of an imidazolidine-based polymer via NSPA based on the imine–yne reaction between 42 and excess 43.37 | |
2.6. Addition–condensation reaction
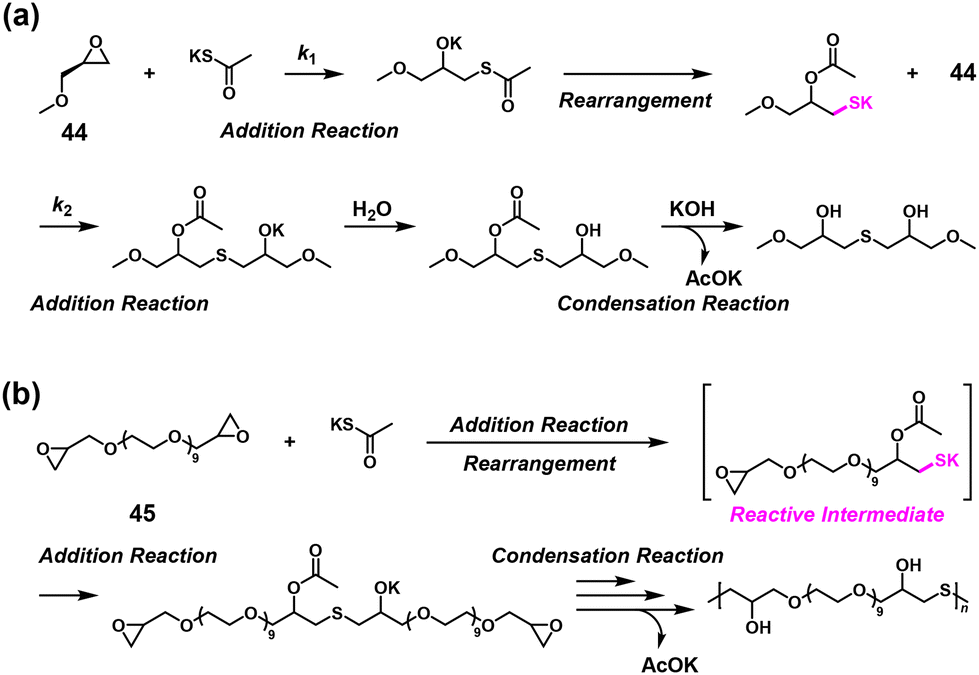
It was found that potassium thioacetate (AcSK) efficiently linked two epoxides in water even under imbalanced stoichiometric ratios.38 The first ring-opening addition reaction between an epoxide compound, (S)-(+)-glycidyl methyl ether (44), and AcSK produced a highly reactive intermediate via a rearrangement that underwent the second ring-opening addition reaction with another 44 with a high κ value of 31, followed by hydrolysis, which eventually condensed potassium acetate (Scheme 20a). The NSPA of a bifunctional epoxide EX-830 (45) with up to 1.6-fold excess of AcSK was achieved, leading to higher molecular weights of the generated poly(thioether)s than calculated using the Carothers/Flory theory (Scheme 20b).
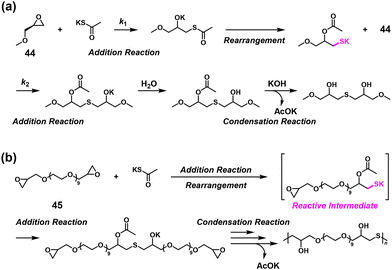 |
| | Scheme 20 (a) Mechanism of the model reaction between 44 and AcSK and (b) synthesis of poly(thioether)s via NSPA based on a thiolate–epoxide reaction between 45 and excess AcSK.38 | |
2.7. Summary
In this section, the strategies for NSSP based on RIM were reviewed in which the B group of the intermediate A–X–Y–B* becomes more reactive than that of the original B–Y–B after the first reaction between the A–X–A and excess B–Y–B monomers to achieve high κ. A wide variety of reactions can be adopted to RIM, including nucleophilic aromatic substitution, electrophilic aromatic substitution, Pd-catalyzed condensation polymerization, ring-closing, polyaddition based on click chemistry, and addition–condensation. One of the most important advantages of NSSP systems is the facile synthesis of high-molecular-weight polymers without strict stoichiometric conditions. For example, the ultrahigh-molecular-weight polymer (Mn = 1140000) can be synthesized by NSSP based on the superacid-catalyzed polyhydroxyalkylation reaction, which is a typical electrophilic aromatic substitution reaction.8 Polyaddition reactions based on click chemistry should also be highlighted in NSSP due to their intrinsically quantitative reaction yields based on low environmental load systems, enabling the synthesis of high-molecular-weight polymers with Mn > 105 without producing any condensates.29–32
3. Intramolecular catalyst transfer system
The ICT system was introduced for realizing chain-growth condensation polymerization (CGCP) of thiophene-based monomers via Kumada–Tamao cross-coupling reactions in the presence of nickel catalysts.40 The studies on CGCP systems have been well reviewed elsewhere.41–45 As mentioned in the Introduction, the ICT system can also be exploited to realize NSSP. According to Fig. 1b, OA of M0 first activates B–Y–B to afford intermediate B*–MII–Y–B. The intermediate reacts with A–X–A via TM or NR to afford intermediate A–X–MII–Y–B. Next, MII intramolecularly transfers to the next B group to afford the activated A–X–Y–MII–B*. Finally, A–X–Y–MII–B* readily reacts with A–X–A in the presence of excess B–Y–B followed by RE to afford A–X–Y–X–A, with the regeneration of M0.
3.1. Palladium-catalyzed condensation polymerization
3.1.1. Tsuji–Trost reaction.
As mentioned in section 2.3, Pd-catalyzed NSCP was achieved based on the cascade bidirectional allylation mechanism to synthesize poly(alkylene vinylene)s.15 A “remote” cascade double allylation strategy was also reported to generate the NSCP system based on the Tsuji–Trost reaction, extending the capable electrophilic (used in excess) and nucleophilic monomers (Fig. 4).46 Importantly, the polymerization mechanism was proposed, which included the ICT system of a Pd0 catalyst after the first allylation reaction (Scheme 21).
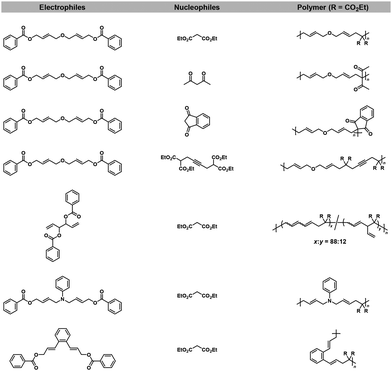 |
| | Fig. 4 Chemical structures of electrophiles and nucleophiles adaptable for NSCP based on the remote cascade double allylation strategy using the Tsuji–Trost reaction.46 | |
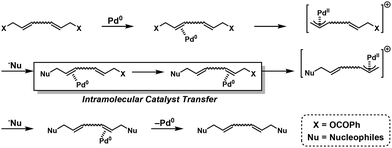 |
| | Scheme 21 The proposed mechanism of NSCP based on the ICT system using the Tsuji–Trost reaction.46 | |
3.1.2. Mizoroki–Heck reaction.
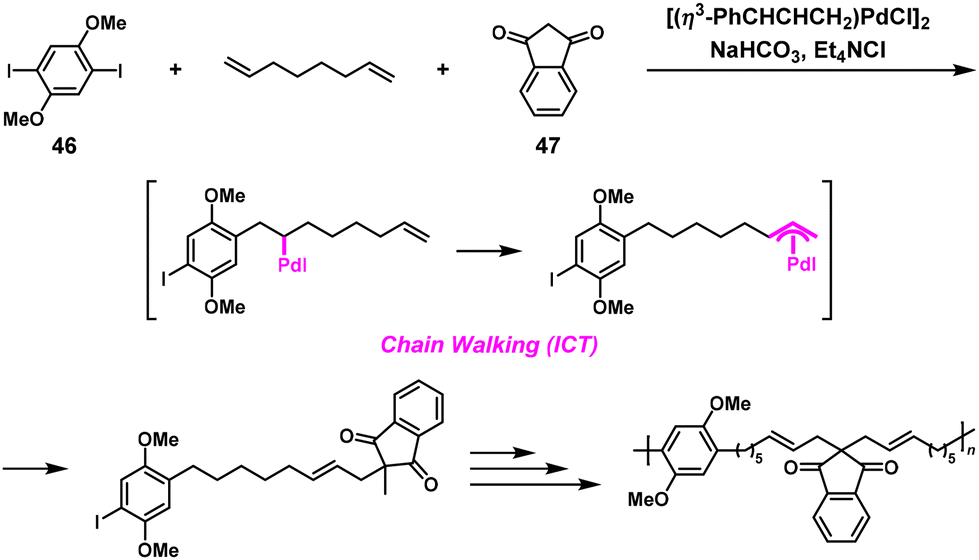
Recently, poly(arylene alkenylene)s were synthesized via NSCP based on the Pd-catalyzed three-component coupling condensation polymerization of diiodoarenes, non-conjugated dienes, and nucleophiles. The polymerization involves the Mizoroki–Heck reaction and subsequent addition of the carbonucleophile to the resulting π-allylpalladium species based on the ICT system.47 Indeed, the condensation polymerization of 1,4-diiodo-2,5-dimethoxybenzene (46), 1,7-octadiene, and 1,3-indandione (47) (1.2-fold excess toward 46) proceeded without issues, yielding poly(arylene alkenylene) with an Mn value of 4700, which was comparable to that obtained under stoichiometric reaction conditions (Mn = 5060) (Scheme 22).
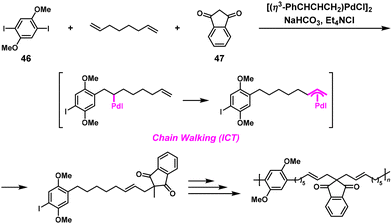 |
| | Scheme 22 Synthesis of poly(arylene alkenylene) via Pd-catalyzed three-component NSCP of 46, 1,7-octadiene, and 47 used in 1.2-fold excess toward 46.47 | |
3.1.3. Migita–Kosugi–Stille cross-coupling reaction.
Our group has proposed the ICT system to achieve NSCP for synthesizing π-conjugated polymers.49 We found that the reaction of an electron-deficient naphthalene–diimide (NDI)-based dibromo monomer (48) and an equimolar amount of 2-(tributylstannyl)thiophene (49) (an excess condition of bromoaryl groups ([Br]0/[Sn]0 = 2)) afforded only a disubstituted compound (50) and unreacted 48, without producing the monosubstituted compound (51) (Scheme 23a). Inspired by the report on NSCP based on the ICT system,46 we proposed that the aforementioned phenomenon may be utilized for NSCP of 1,3-bis(trimethylstannyl)thiophene (52) and excess 48 (Scheme 23b).
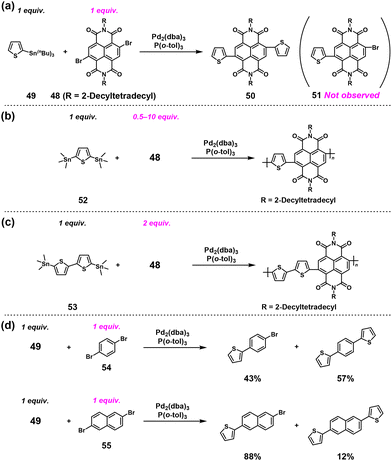 |
| | Scheme 23 (a) The reaction between 49 and an equimolar amount of 48, (b) synthesis of an NDI-based N-type semiconducting polymer via NSCP of 52 with excess 48 based on a Migita–Kosugi–Stille cross-coupling reaction, (c) synthesis of an NDI-based N-type semiconducting polymer via NSCP of 53 with excess 48 based on a Migita–Kosugi–Stille cross-coupling reaction, and (d) model reactions between 49 and an equimolar amount of 54/55.49 | |
Indeed, NSCP based on the Migita–Kosugi–Stille coupling reaction of 52 with up to 10-fold excess of 48 produced NDI-based N-type semiconducting polymers with much higher Mn values than theoretical values based on eqn (2) (Table 1). Using 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (53, Scheme 23c) instead of 52 also proved to be successful for synthesizing another high-molecular-weight NDI-based N-type semiconducting polymer (Mn = 22600) via NSCP, demonstrating the generality of the proposed ICT system.
Table 1 Results of NSCP between 52 and excess 48 based on the Migita–Kosugi–Stille cross-coupling reaction49
| [52]0:[48]0 |
M
n (SEC) |
M
n (theory) |
M
w/Mn (SEC) |
| 2:1 |
— |
1530 |
— |
| 1:1 |
23500 |
— |
2.00 |
| 1:1.25 |
17800 |
4590 |
1.93 |
| 1:2 |
14500 |
1530 |
1.64 |
| 1:4 |
10200 |
850 |
1.46 |
| 1:10 |
8500 |
623 |
1.42 |
To determine where these ICT phenomena originate, the model reaction of 49 with an equimolar amount of 1,4-dibromobenzene (54) or 2,6-dibromonaphthalene (55), which are dibromo compounds without imide groups, was performed under the condition of [Br]0/[Sn]0 = 2 (Scheme 23d). Monosubstituted products were obtained in 43% and 88% yields using 54 and 55, respectively, and the selectivity for forming disubstituted products was greatly reduced. Thus, the presence of an imide group in a dibromo monomer was important for the occurrence of ICT phenomena.
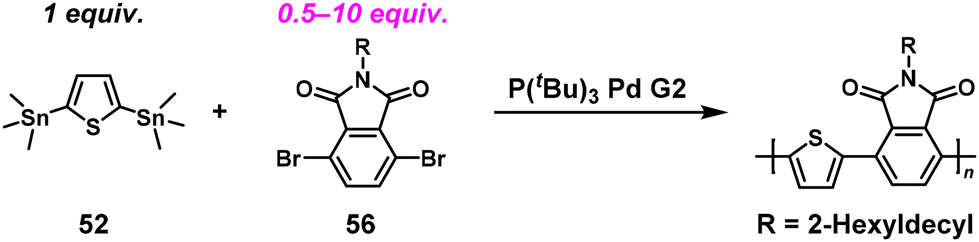
Based on these findings, we newly designed a phthalimide derivative (56) as a monoimide dibromo compound and conducted ICT-assisted NSCP based on the Migita–Kosugi–Stille coupling reaction ([56]0/[52]0 = 2–10) (Scheme 24).50 A phthalimide-containing π-conjugated polymer was obtained with higher Mn values than predicted (Table 2). Notably, the catalysts had a substantial effect in this system; the use of a chloro[(tri-tert-butylphosphine)-2-(2-aminobiphenyl)] palladium(II) (P(tBu)3Pd G2) produced high-molecular-weight polymers under nonstoichiometric conditions.
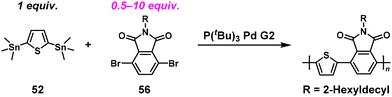 |
| | Scheme 24 Synthesis of phthalimide-containing π-conjugated polymers via NSCP of 52 with excess 56 based on the Migita–Kosugi–Stille cross-coupling reaction.50 | |
Table 2 Results of NSCP between 52 and excess 56 based on the Migita–Kosugi–Stille cross-coupling reaction50
| [52]0:[56]0 |
M
n (SEC) |
M
n (theory) |
M
w/Mn (SEC) |
| 2:1 |
— |
1400 |
— |
| 1:1 |
22300 |
— |
2.30 |
| 1:2 |
16000 |
1400 |
2.51 |
| 1:4 |
10000 |
750 |
2.51 |
| 1:10 |
5900 |
550 |
1.82 |
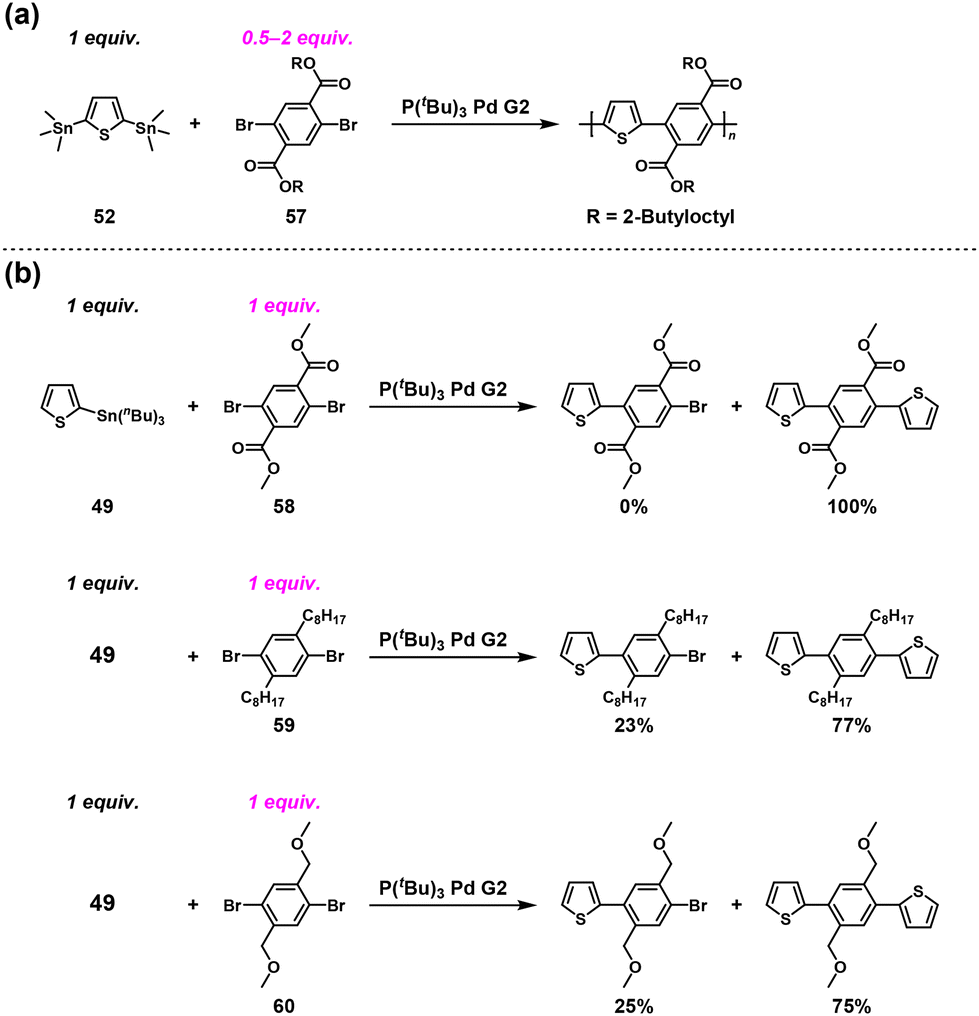
Furthermore, we wanted to determine whether imide groups are essential for the ICT-assisted NSCP. Because we speculated the importance of carbonyl groups introduced at the ortho positions of dibromo monomers, an ester-containing dihalo monomer (57) was adopted as an alternative in a similar polymerization system.51 NSCP of 52 with a 2-fold excess of 57 was performed to synthesize an ester-containing π-conjugated polymer in the presence of P(tBu)3Pd G2 (Scheme 25a). The observed Mn value of the resulting polymers was 16800, which was much higher than expected (Mtheoryn = 874). Thus, the ICT system originates from an ortho carbonyl group substituted in dibromo monomers, indicating that “imide” groups are not a crucial molecular design element for dibromo monomers.
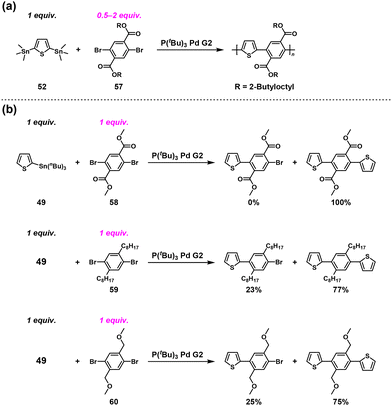 |
| | Scheme 25 (a) Synthesis of ester-containing π-conjugated polymers via NSCP of 52 with excess 57 based on the Migita–Kosugi–Stille cross-coupling reaction and (b) model reactions between 49 and an equimolar amount of 58/59/60.51 | |
To investigate the origin of the ICT phenomenon, the model reactions between 49 and an equimolar amount of dimethyl 2,5-dibromoterephthalate (58), 1,4-dibromo-2,5-dioctylbenzene (59), or 1,4-dibromo-2,5-bis(methoxymethyl)benzene (60) ([Br]0/[Sn]0 = 2) were investigated in detail (Scheme 25b). Monosubstituted compounds were obtained in 0%, 23%, and 25% yields using 58, 59, and 60, respectively, supporting the necessity of ortho carbonyl groups in the dibromo compounds to achieve ICT-assisted NSCP based on the Migita–Kosugi–Stille cross-coupling reaction. In addition, the density functional theory calculation results indicated that the intermediate compound generated, Ar–PdII–Br (Ar = aryl) with PdII/CO association, was much more stable than its other counterparts by 9–10 kcal mol−1. This would be one of the driving forces of the ICT system.51 We speculated that the ortho carbonyl groups form a pseudo-5-membered ring with PdII, which considerably stabilizes the intermediate state (Fig. 5).
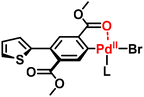 |
| | Fig. 5 The proposed intermediate with a pseudo-5-membered ring generated via the model reaction between 49 and 58.51 | |
The mechanism of the ICT-assisted NSCP based on the Migita–Kosugi–Stille coupling reaction is summarized in Scheme 2651 as follows. (a) OA of Pd0 with the dibromo monomer, Br–Ar–Br, used in excess produces Br–PdII–Ar–Br, (b) TM occurs between Br–PdII–Ar–Br and the incoming distannylated compounds, Me3Sn–Pn–Ar′–SnMe3 (Pn = polymer with a DPn of n), to afford Me3Sn–Pn–Ar′–PdII–Ar–Br, (c) RE of PdII affords an associated pair of Pd0 and the generated Me3Sn–Pn+1–Br via Pd0-π interactions (first cross-coupling reaction), (d) Pd0 intramolecularly transfers to the other side of the Ar–Br group to form Me3Sn–Pn+1–PdII–Br with a pseudo-5-membered ring via OA, (e) TM between Me3Sn–Pn+1–PdII–Br and the other incoming distannylated compounds, Me3Sn–Pm–Ar′–SnMe3, affords Me3Sn–Pn+1–PdII–Pm–Ar′–SnMe3, and (f) Pd0 regenerates via RE to obtain Me3Sn–Pn+m+1–Ar′–SnMe3 (second cross-coupling reaction). During this catalytic cycle, the generating oligomers are always end-capped with stannyl groups; only a stoichiometric amount of dibromo monomers can participate in the polymerization to achieve NSCP.
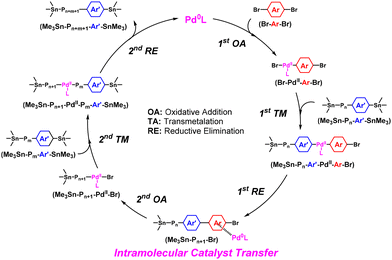 |
| | Scheme 26 The proposed mechanism of the ICT-assisted NSCP based on the Migita–Kosugi–Stille coupling reaction. | |
3.1.4. Suzuki–Miyaura cross-coupling reaction.
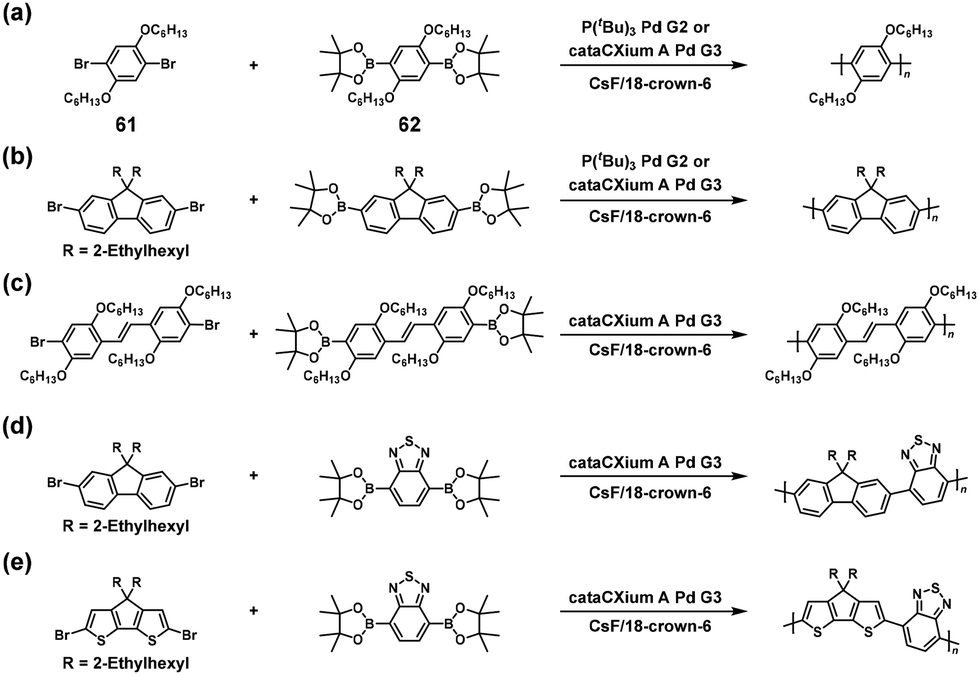
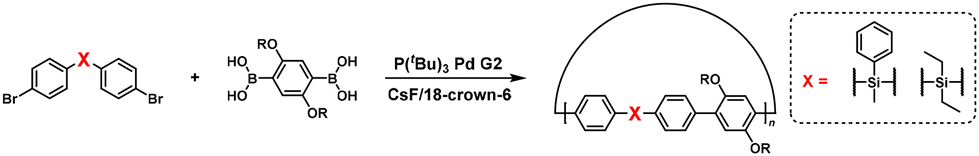
NSCP was achieved based on the Suzuki–Miyaura cross-coupling reaction via the ICT system. Indeed, high-molecular-weight poly(p-phenylene) (Mn = 19400, Mw/Mn = 2.55) was obtained via NSCP of a phenylene-based diboronic acid ester monomer (62) with a 1.3-fold excess of a phenylene-based dibromo monomer (61) in the presence of P(tBu)3Pd G2 (Scheme 27a).52 Furthermore, Pd cataCXium was more effective than P(tBu)3Pd G2 for obtaining fluorene- and cyclopentadithiophene-containing π-conjugated polymers with Mn values of > 10000 (Scheme 27b–e).55
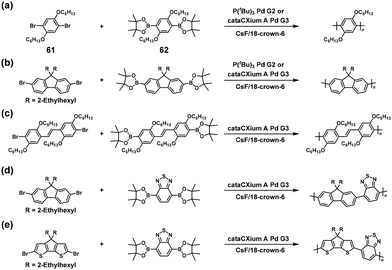 |
| | Scheme 27 Synthesis of π-conjugated polyarylenes via NSCP based on the Suzuki–Miyaura cross-coupling reaction of diboronic acid ester monomers with excess dibromo monomers. Synthesis of (a) polyphenylene, (b) polyfluorene, (c) polystilbene, (d) poly(fluorene-alt-benzothiadiazole), and (e) poly(cyclopentadithiophene-alt-benzothiadiazole).52,55 | |
It was explained that a dibromo monomer with electron-rich donor units outperforms one with electron-deficient units in NSCP because of the strong affinity of the donor aromatic for the Pd catalyst.52 Furthermore, the α,ω-chain-end groups of the obtained polymer primarily corresponded to pinacol boronate groups despite using an excess of dibromo monomers, as confirmed by using matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry, supporting the ICT system.
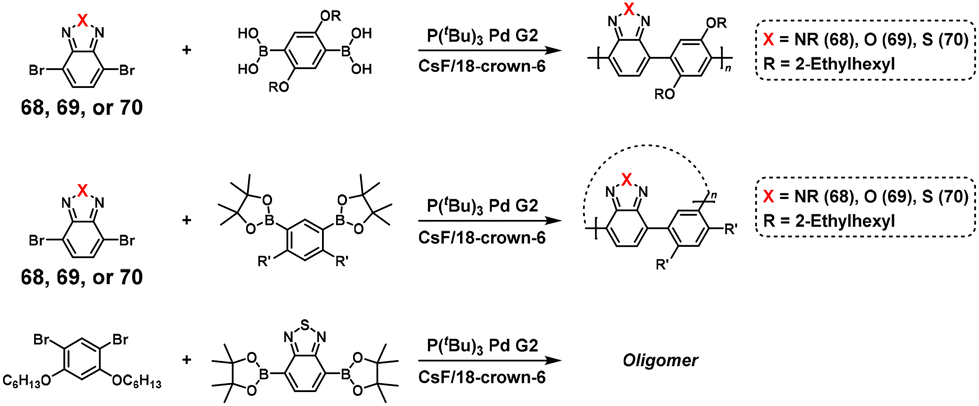
The NSCP of diboronic acid pinacol ester monomers (63 and 64) with excess dibromo monomers with CC (65),53 C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (66),54 and NN (67)54 bonds was also reported based on the ICT system (Scheme 28a–c). The authors also investigated the ICT phenomena in detail on a variety of functional groups of diphenylene dibromo monomers (BrPh–X–PhBr), demonstrating NSCP between phenylenediboronic acid ester and a 1.3-fold excess of BrPh–X–PhBr.56 As a result, high-molecular-weight polymers were obtained for X = CH2, CO, N–Bu, O, and SO2 (Scheme 28d). Furthermore, cyclic polymers were formed using BrPh–X–PhBr (X = CH2, N–Bu, and O).56
C (66),54 and NN (67)54 bonds was also reported based on the ICT system (Scheme 28a–c). The authors also investigated the ICT phenomena in detail on a variety of functional groups of diphenylene dibromo monomers (BrPh–X–PhBr), demonstrating NSCP between phenylenediboronic acid ester and a 1.3-fold excess of BrPh–X–PhBr.56 As a result, high-molecular-weight polymers were obtained for X = CH2, CO, N–Bu, O, and SO2 (Scheme 28d). Furthermore, cyclic polymers were formed using BrPh–X–PhBr (X = CH2, N–Bu, and O).56
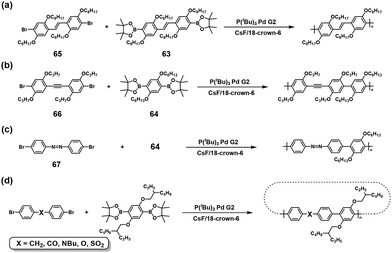 |
| | Scheme 28 Synthesis of π-conjugated polyarylenes via NSCP based on the Suzuki–Miyaura cross-coupling reaction between diboronic acid ester monomers and excess dibromo monomers with various functional groups. Synthesis of polyarylenes with (a) vinylene, (b) alkynylene, (c) diazo, and (d) various functional spaser units in main chains.53,54,56 | |
The NSCP of phenylene-based diboronic acid esters with a 1.3–2.0-fold excess of N-acyl dithieno[3,2-b:2′,3′-d]pyrroles was also demonstrated to successfully synthesize π-conjugated polymers with ultrahigh Mn values up to 195000 (Scheme 29).57 The use of tripotassium phosphate as a base was essential for increasing the polymerization rate.
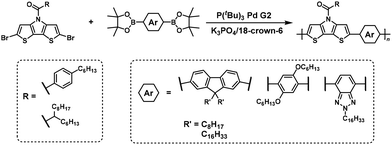 |
| | Scheme 29 Synthesis of π-conjugated polyarylenes via NSCP based on the Suzuki–Miyaura cross-coupling reaction of various diboronic acid ester monomers with excess N-acyl dithieno[3,2-b:2′,3′-d]pyrroles.57 | |
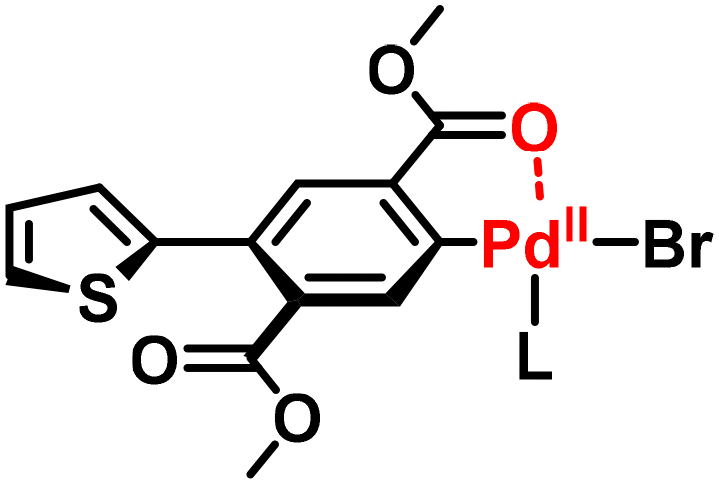
Furthermore, the NSCP and related model reactions were performed using diboronic acid esters and excess dibromobenzotriazole (68), dibromobenzoxazole (69), or dibromobenzothiadiazole (70) (Scheme 30).58 The ICT phenomenon was observed when using 68, whereas the use of 69 resulted in incomplete ICT with a concurrent intermolecular Pd transfer system between any dibromo monomers. The use of 70 resulted in a dominant intermolecular Pd transfer system. Thus, the molecular design of dibromo monomers is important for realizing NSCP based on the Suzuki–Miyaura cross-coupling reaction.
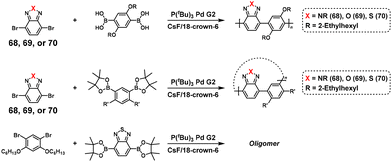 |
| | Scheme 30 Synthesis of polyarylenes via NSCP based on the Suzuki–Miyaura cross-coupling reaction between diboronic acid ester monomers and excess 68, 69, or 70.58 | |
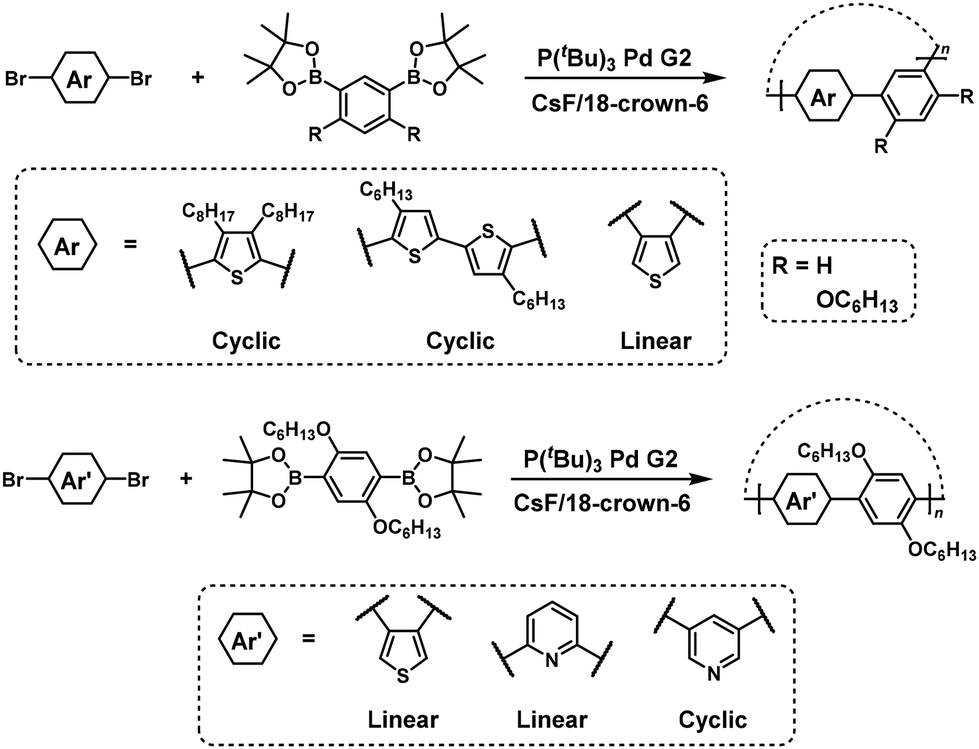
Cyclic polymers were predominantly synthesized via NSCP of m-phenylene diboronic acid ester (71) with a 1.3-fold excess of p-dibromobenzene (72) in the presence of P(tBu)3Pd G2, as confirmed using MALDI-TOF mass spectrometry (Scheme 31).59 The authors explained that an excess of a dibromo monomer should promote the generation of the intermediate formation of a Br–Pd/boronic acid pinacol (Bpin)-ended polymer in the final stage, exclusively producing a cyclic polymer. In addition, a kinked structure based on meta-substituted dibromo- or diboronic acid ester monomers is essential for the chemoselective formation of cyclic polymers.59,60
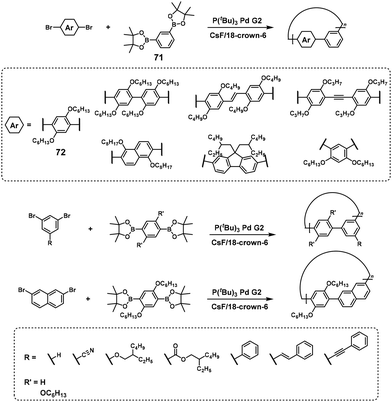 |
| | Scheme 31 Synthesis of cyclic polyarylenes via NSCP based on the Suzuki–Miyaura cross-coupling reaction.59,60 | |
Dibromo heteroarylene monomers can also be used to synthesize cyclic π-conjugated polymers via NSCP (Scheme 32).61 Again, the position of bromo substituents in dibromo heteroarylene monomers plays an important role in whether cyclic or linear polymers are formed. As an exception, para-substituted dibromo monomers with a silylene group can participate in NSCP based on the Suzuki–Miyaura cross-coupling reaction to afford cyclic π-conjugated polymers (Scheme 33).62 The authors explained that sp3-silicon should induce bent structures, thereby promoting the formation of cyclic polymers.
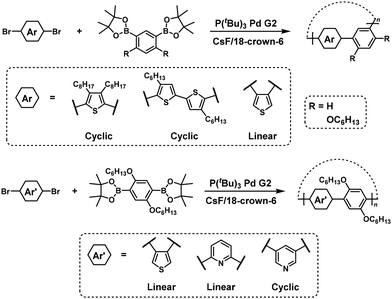 |
| | Scheme 32 Synthesis of cyclic poly(heteroarylene)s via NSCP based on the Suzuki–Miyaura cross-coupling reaction.61 | |
 |
| | Scheme 33 Synthesis of cyclic poly(heteroarylene)s via NSCP based on the Suzuki–Miyaura cross-coupling reaction.62 | |
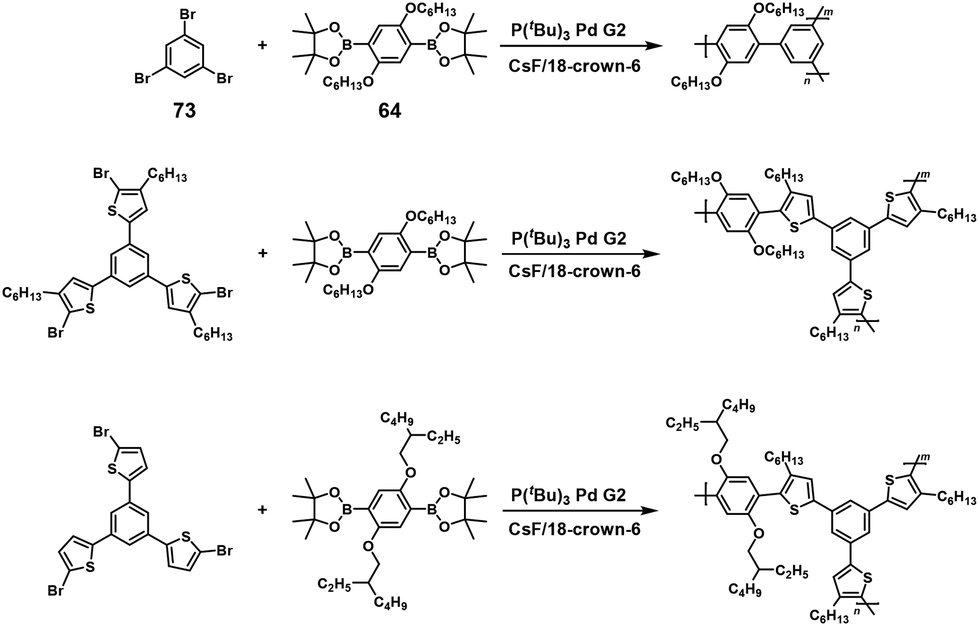
Recently, soluble high-molecular-weight hyperbranched polyphenylenes with a 100% degree of branching (DB) were synthesized via A3 + B2 type NSCP based on the Suzuki–Miyaura cross-coupling reaction using 64 and a 1.0–3.0-fold excess of 1,3,5-tribromobenzene (73) in the presence of P(tBu)3Pd G2 (Scheme 34).63 The obtained polymer (under a stoichiometric condition of [Br]0/[B]0 = 1) had a DPn of 83.5, which was much higher than the value (6.6) predicted by the Flory–Stockmayer theory. This substantial deviation is explained by the enhanced reactivity of the remaining two bromines in 73 after the first substitution of Br with 64 or BPin of the generated oligomers via ICT. Similarly, high-molecular-weight hyperbranched poly(phenylene thienylene)s with a 100% DB were obtained via A3 + B2 type NSCP based on the Suzuki–Miyaura cross-coupling reaction (see also Scheme 34).64
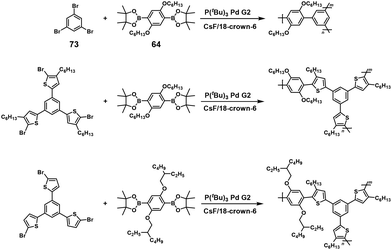 |
| | Scheme 34 Synthesis of hyperbranched polyarylenes with a 100% degree of branching via A3 + B2 type NSCP based on the Suzuki–Miyaura cross-coupling reaction.63,64 | |
3.1.5. Direct arylation polymerization.
An atom-economical approach was proposed to achieve NSCP based on the direct arylation reaction.65 Indeed, the direct arylation polymerization (DArP) of 2,2′,3,3′,5,5′,6,6′-octafluorobiphenyl (74) with excess 2,7-diiodo-9,9-dioctyl-9H-fluorene (75) ([75]0/[74]0 = 1–3) was catalyzed by Pd2(dba)3/tricyclohexylphosphine (PCy3) in the presence of Ag2CO3 (2 equiv.) and K2CO3 (12 equiv.) to obtain high-molecular-weight polyarylenes (Mn = 18000–33000) (Scheme 35). A Pd/silver (Ag) dual catalyst system was proposed in which the catalyst could “walk through” fluorene moieties.
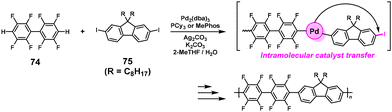 |
| | Scheme 35 Synthesis of polyarylenes via NSCP based on DArP via the Pd/Ag dual catalyst system.65 | |
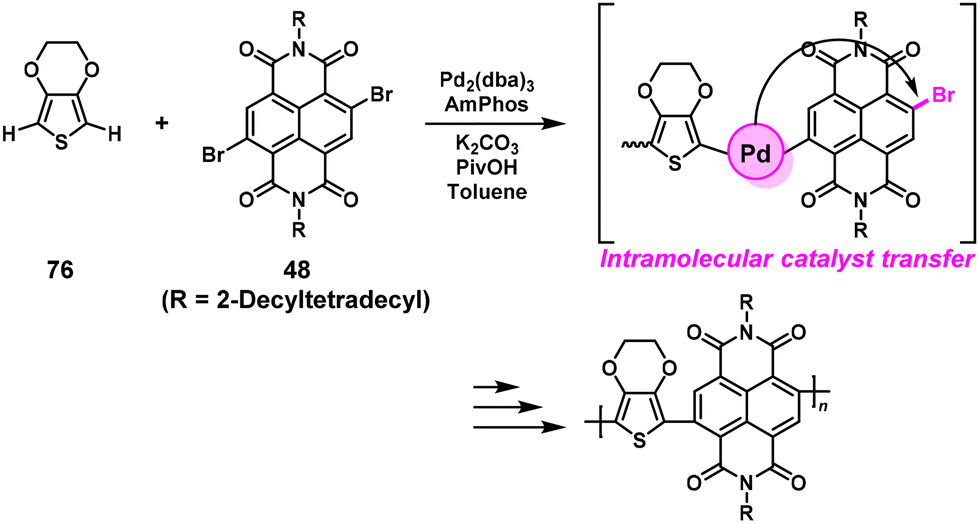
Our group reported preliminary results of NSCP based on DArP catalyzed by a single catalyst of Pd with the (4-dimethylaminophenyl)di-tert-butylphosphine (AmPhos) ligand (Scheme 36).66 In practice, high-molecular-weight poly(naphthalene diimide-alt-ethylenedioxythiophene)s (Mn = 13000–17000) were successfully synthesized via NSCP of 2,3-dihydrothieno[3,4-b][1,4]dioxine (76) with excess 48 ([48]0/[76]0 = 1–3).
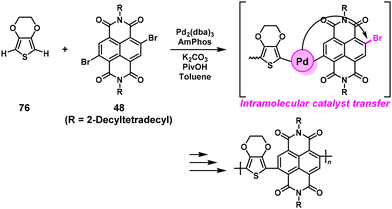 |
| | Scheme 36 Synthesis of polyarylenes via NSCP based on DArP via a Pd single catalyst system.66 | |
3.2. Cobalt-catalyzed polyaddition reaction
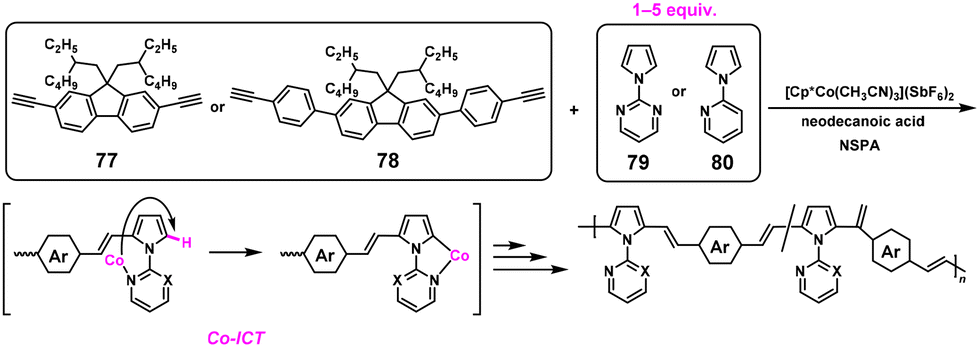
As introduced in section 2.5, the NSPA system is the most atom-economical approach without generating condensates during polymerization. In practice, high-molecular-weight poly(arylene vinylene)s with Mn values of 20000–52000 were obtained via NSPA based on the cobalt (Co)-catalyzed polyaddition reaction. The Co-catalyzed hydroarylation reactions between diyne monomers (77 and 78) and excess 1-(2-pyrimidinyl)pyrrole (79) or 1-(2-pyridyl)pyrrole (80) were performed under nonstoichiometric conditions with monomer ratios up to 1:5 in the presence of [Cp*Co(CH3CN)3](SbF6)2 and neodecanoic acid (NDA, 30 mol%) (Scheme 37).67 The Co ICT system was proposed, walking through the aromatic ring induced by the 2-pyrimidinyl substituent.
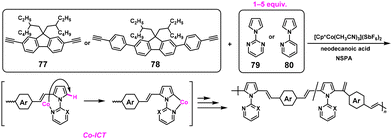 |
| | Scheme 37 Synthesis of poly(arylene vinylene)s via NSPA based on the Co-ICT system.67 | |
3.3. Rhodium-catalyzed condensation polymerization
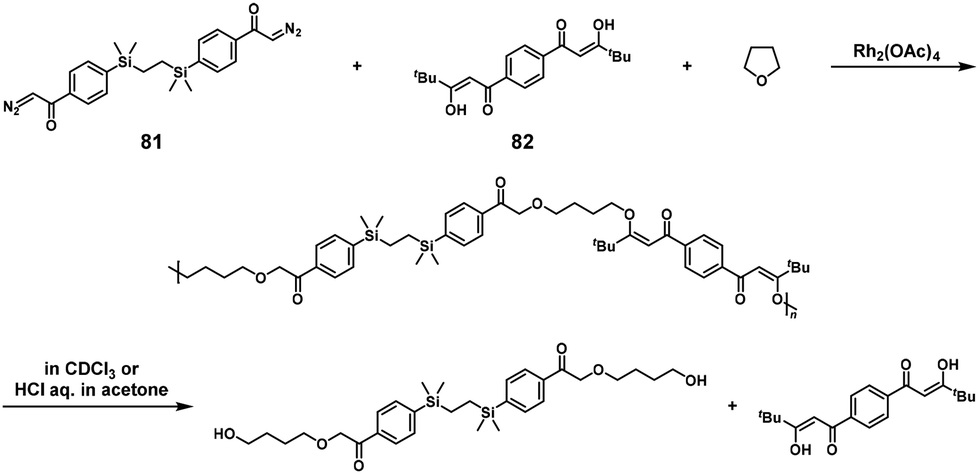
The synthesis of poly(β-keto enol ether)s was performed via Rh-catalyzed three-component NSCP of bis(diazoketone) (used in excess), bis(1,3-diketone), and tetrahydrofuran. The ICT phenomenon was speculated for realizing NSCP (Scheme 38).48 For example, high-molecular-weight poly(β-keto enol ether) was obtained using a 1.2-fold excess of 1,2-bis[(4-diazoacetylphenyl)dimethylsilyl]ethane (81) toward 1,1′-(1,4-phenylene)bis-3,3-dimethylpentane-1,3-dione (82) (Mn = 42800). The degradation reaction of the obtained polymers was also reported by the same group under acidic conditions to afford well-defined low-molecular-weight components.
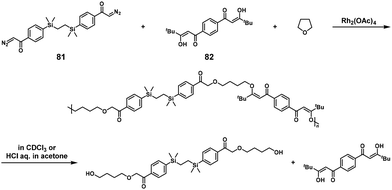 |
| | Scheme 38 Synthesis of poly(β-keto enol ether) via Rh-catalyzed three-component NSCP of bis(diazoketone) (used in excess), bis(1,3-diketone), and tetrahydrofuran, and degradation of poly(β-keto enol ether) under acidic conditions.48 | |
3.4. Summary
In this section, the strategies for NSSP based on the unique ICT systems were reviewed in which a transition metal catalyst walked through one of the monomers to accelerate the second reaction after the first one. The ICT-assisted NSSP systems are found in Pd-catalyzed condensation polymerization (Tsuji–Trost, Mizoroki–Heck, Migita–Kosugi–Stille, Suzuki–Miyaura cross-coupling polymerization, and direct arylation polymerization), Co-catalyzed polyaddition, and Rh-catalyzed condensation polymerization. In particular, an authentic polymerization method based on the Suzuki–Miyaura cross-coupling reaction can increase Mn values up to 195000 even under an imbalanced monomer feed ratio.57 On the other hand, the NSSP systems based on DArP are especially interesting because of their advantages of environmental friendliness, having the possibility to be commercialized by employing metal-free monomers prepared via reduced reaction steps.
4. Conclusions
This paper reviews NSSP systems, including NSCP and NSPA, focusing on recent progress in their use for synthesizing high-molecular-weight polymers. The RIM and ICT mechanisms are essential driving forces to achieve efficient NSSP, resulting in the generation of polymers with much higher Mn values than expected from the theory established by Carothers and Flory. The advantages of the NSSP systems are as follows. (a) Strict balancing of feed ratios of A–X–A and B–Y–B monomers is unnecessary. (b) Exact stoichiometry of actual functionalities of monomers in polymerization is unnecessary, even under conditions such as side reactions, volatile monomers, and precipitation of generated polymers. (c) The polymerization rate is increased by increasing the concentration of one of the monomers. (d) The functional group at the chain ends of the resulting polymer (A–polymer–A or B–polymer–B) is controllable, making it suitable as a starting polymer for synthesizing well-defined triblock or multiblock copolymers. The origins of the RIM and ICT phenomena have been investigated in NSSP based on various reactions to some extent. However, the successes in NSSP are still limited to certain substrates of monomers, and the general molecular design strategies have not yet been established. Expanding the range of adaptable monomers would generalize and establish each NSSP system, which is essential for creating new functional polymer materials suited for a wide variety of specific applications. The screening of the adaptable monomers would be streamlined using AI, machine learning, etc.
Author contributions
KY contributed to writing – original draft and TH contributed to writing – review & editing. All authors reviewed the results and approved the final version of the manuscript.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Management Expense Grant. No. 39621347E0, Yamagata University.
References
- W. H. Carothers, Trans. Faraday Soc., 1936, 32, 39–49 RSC
 .
.
- P. J. Flory, J. Am. Chem. Soc., 1936, 58, 1877–1885 CAS .
- P. J. Flory, Chem. Rev., 1946, 39, 137–197 CAS .
- A. S. Hay, F. J. Williams, H. M. Relies and B. M. Boulette, J. Macromol. Sci., Part A:Pure Appl. Chem., 1984, 21, 1065–1079 Search PubMed .
- K. Miyatake, A. R. Hill and A. S. Hay, Macromolecules, 2001, 34, 4288–4290 CAS .
- N. Kihara, S. Komatsu, T. Takata and T. Endo, Macromolecules, 1999, 32, 4776–4783 CAS .
- H. Iimori, Y. Shibasaki, S. Ando and M. Ueda, Macromol. Symp., 2003, 199, 23–36 CrossRef CAS .
- M. T. Guzmán-Gutiérrez, D. R. Nieto, S. Fomine, S. L. Morales, M. G. Zolotukhin, M. C. G. Hernandez, H. Kricheldorf and E. S. Wilks, Macromolecules, 2011, 44, 194–202 CrossRef .
- A. R. Cruz, M. C. G. Hernandez, M. T. Guzmán-Gutiérrez, M. G. Zolotukhin, S. Fomine, S. L. Morales, H. Kricheldorf, E. S. Wilks, J. Cárdenas and M. Salmón, Macromolecules, 2012, 45, 6774–6780 CrossRef CAS .
- L. I. Olvera, M. G. Zolotukhin, O. Hernández-Cruz, S. Fomine, J. Cárdenas, R. L. Gaviño-Ramírez and F. A. Ruiz-Trevino, ACS Macro Lett., 2015, 4, 492–494 CrossRef CAS .
- K. Matsumoto, T. Ogawa and M. Jikei, Polym. Chem., 2017, 8, 7297–7300 RSC .
- K. Matsumoto, C. Fukui, R. Shoji and M. Jikei, Polym. Chem., 2020, 11, 4221–4227 RSC .
- H. C. Kiliclar, E. Gencosman and Y. Yagci, Macromol. Rapid Commun., 2022, 43, 2100584 CAS .
- E. Gencosman, H. C. Kiliclar, P. Fiedor, G. Yilmaz, J. Ortyl, Y. Yagci and B. Kiskan, Macromol. Rapid Commun., 2024, 45, 2300458 CAS .
- N. Nomura, K. Tsurugi and M. Okada, Angew. Chem., Int. Ed., 2001, 40, 1932–1935 CAS .
- T. Koizumi, K. Sugie, O. Kiyonaga, M. Yamanaka and S. Kawabata, Macromolecules, 2004, 37, 9670–9672 CAS .
- T. Takemura, K. Sugie, H. Nishino, S. Kawabata and T. Koizumi, J. Polym. Sci., Part A:Polym. Chem., 2008, 46, 2250–2261 CrossRef CAS .
- N. Nishioka and T. Koizumi, Eur. Polym. J., 2011, 47, 1142–1150 CrossRef CAS .
- H. Nishino, N. Nishioka and T. Koizumi, Polym. J., 2012, 44, 321–326 CrossRef CAS .
- N. Nishioka, S. Hayashi and T. Koizumi, Angew. Chem., Int. Ed., 2012, 51, 3682–3685 CrossRef CAS PubMed .
- M. Gao, J. W. Y. Lam, J. Li, C. Y. K. Chan, Y. Chen, N. Zhao, T. Han and B. Z. Tang, Polym. Chem., 2013, 4, 1372–1380 RSC .
- M. Gao, J. W. Y. Lam, Y. Liu, J. Li and B. Z. Tang, Polym. Chem., 2013, 4, 2841 RSC .
- Y. Liu, M. Gao, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2016, 7, 5436–5444 RSC .
- T. Han, Z. Zhao, H. Deng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2017, 8, 1393–1403 CAS .
- J. Zhang, W. Wang, Y. Liu, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2017, 50, 8554–8561 CrossRef CAS .
- T. Han, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2018, 9, 885–893 RSC .
- Q. Gao, T. Han, Z. Qiu, R. Zhang, J. Zhang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2019, 10, 5296–5303 RSC .
- J.-P. Lindner, Macromolecules, 2016, 49, 2046–2053 CrossRef CAS .
- J.-Q. Chen, L. Xiang, X. Liu, X. Liu and K. Zhang, Macromolecules, 2017, 50, 5790–5797 CAS .
- L. Xiang, Z. Li, J. Liu, J. Chen, M. Zhang, Y. Wu and K. Zhang, Polym. Chem., 2018, 9, 4036–4043 RSC .
- L. Zhang, X. Ren, Y. Zhang and K. Zhang, ACS Macro Lett., 2019, 8, 948–954 CrossRef CAS PubMed .
- X. Liu, L. Xiang, J. Li, Y. Wu and K. Zhang, Polym. Chem., 2020, 11, 125–134 CAS .
- Q. Tang and K. Zhang, Chin. J. Chem., 2021, 39, 3093–3100 CrossRef CAS .
- J.-S. Wu, W.-M. Hou, Z.-J. Liu, Y. Shi and Y.-M. Chen, Chin. J. Polym. Sci., 2023, 41, 760–767 CrossRef CAS .
- Y. Zhang, Q. Tang, Z. Li and K. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202302527 CrossRef CAS .
- Y. Zhang, J. Zheng, X. Li, Y. Wu and K. Zhang, Chem. Res. Chin. Univ., 2023, 39, 822–828 CrossRef CAS .
- J. Chen, X. Xu, Q. Xia, J. Wang, A. Qin and B. Z. Tang, Macromolecules, 2024, 57, 1970–1978 CrossRef CAS .
- T. Kishida, T. Shimada and K. Sugiyasu, Chem. Commun., 2022, 58, 1108–1110 RSC .
- T. Dutta, K. B. Woody and M. D. Watson, J. Am. Chem. Soc., 2008, 130, 452–453 CrossRef CAS .
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542–17547 CrossRef CAS PubMed .
- T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595–5619 CrossRef CAS PubMed .
- T. Yokozawa and Y. Ohta, Chem. Rev., 2016, 116, 1950–1968 CrossRef CAS .
- M. P. Aplan and E. D. Gomez, Ind. Eng. Chem. Res., 2017, 56, 7888–7901 CrossRef CAS .
- A. K. Leone, P. K. Goldberg and A. J. McNeil, J. Am. Chem. Soc., 2018, 140, 7846–7850 CrossRef CAS PubMed .
- C. N. Jarrett-Wilkins, A. A. Pollit and D. S. Seferos, Trends Chem., 2020, 2, 493–505 CrossRef CAS .
- N. Nomura, K. Tsurugi, T. V. RajanBabu and T. Kondo, J. Am. Chem. Soc., 2004, 126, 5354–5355 CrossRef CAS PubMed .
- L. Tan, D. Takeuchi and K. Osakada, J. Polym. Sci., Part A:Polym. Chem., 2019, 57, 2535–2542 CrossRef CAS .
- H. Shimomoto, T. Mori, T. Itoh and E. Ihara, Macromolecules, 2019, 52, 5761–5768 CrossRef CAS .
- E. Goto, S. Ando, M. Ueda and T. Higashihara, ACS Macro Lett., 2015, 4, 1004–1007 Search PubMed .
- K. Terayama, E. Goto and T. Higashihara, Macromol. Chem. Phys., 2018, 219, 1800175 Search PubMed .
- K. Terayama, C.-W. Liu and T. Higashihara, Polym. J., 2022, 54, 143–150 CAS .
- M. Nojima, K. Kosaka, M. Kato, Y. Ohta and T. Yokozawa, Macromol. Rapid Commun., 2016, 37, 79–85 CAS .
- M. Nojima, Y. Ohta and T. Yokozawa, Chem. Lett., 2017, 46, 35–37 Search PubMed .
- T. Kamigawara, H. Sugita, K. Mikami, Y. Ohta and T. Yokozawa, Catalysts, 2017, 7, 195 Search PubMed .
- K. Kosaka, K. Nanjyo, Y. Ohta and T. Yokozawa, Chem. Lett., 2018, 47, 1040–1043 Search PubMed .
- T. Yokozawa, N. Harada, H. Sugita and Y. Ohta, Chem. – Eur. J., 2019, 25, 10059–10062 CAS .
- T. H. Nguyen, L.-T. T. Nguyen, M. H. Hoang, T.-Q. Nguyen, S. T. Cu, R. Simada, Y. Ohta, T. Yokozawa and H. T. Nguyen, Eur. Polym. J., 2023, 186, 111867 CAS .
- H. Sugita, T. Kamigawara, S. Miyazaki, R. Shimada, T. Katoh, Y. Ohta and T. Yokozawa, Chem. – Eur. J., 2023, 29, e202301242 CAS .
- H. Sugita, M. Nojima, Y. Ohta and T. Yokozawa, Chem. Commun., 2017, 53, 396–399 Search PubMed .
- H. Sugita, M. Nojima, Y. Ohta and T. Yokozawa, Polym. Chem., 2019, 10, 1182–1185 RSC .
- H. Sugita, Y. Ohta and T. Yokozawa, J. Polym. Sci., 2020, 58, 1236–1240 CrossRef CAS .
- N. Harada, R. Yachida, R. Shimada, T. Katoh, S. Hashimoto, Y. Ohta, I. Iwakura and T. Yokozawa, Polym. J., 2024, 56, 777–781 CrossRef CAS .
- R. Shimada, Y. Takahashi, H. Sugita, Y. Ohta and T. Yokozawa, Chem. Lett., 2023, 52, 665–668 CrossRef CAS .
- R. Shimada, Y. Ohta and T. Yokozawa, Eur. Polym. J., 2024, 214, 113155 CrossRef CAS .
- L. Takimoto, T. Goto, J. Chen, J. Kuwabara and T. Kanbara, Macromol. Rapid Commun., 2024, 45, 2300225 CrossRef CAS PubMed .
-
K. Yokawa and T. Higashihara, Proceedings in 31stJapan Polyimide & Aromatic Polymers Conference, 2024, 31, 185–188.
- R. Iwamori, R. Sato, J. Kuwabara and T. Kanbara, Polym. Chem., 2022, 13, 379–382 RSC .
- D. Zhao and J. S. Moore, J. Am. Chem. Soc., 2003, 125, 16294–16299 CrossRef CAS PubMed .
- D. Zhao and K. Yue, Macromolecules, 2008, 41, 4029–4036 CrossRef CAS .
- K. Kimura, S. Kohama and Y. Yamashita, Macromolecules, 2002, 35, 7545–7552 CrossRef CAS .
- K. Kimura, S. Kohama and Y. Yamashita, Macromolecules, 2003, 36, 5043–5046 CrossRef CAS .
- S. Kohama, K. Kimura, T. Uchida, S. Umehara, Y. Ikemoto and Y. Yamashita, Polym. Int., 2005, 54, 471–480 CrossRef CAS .
- S. Kohama, K. Kimura and Y. Yamashita, J. Polym. Sci., Part A:Polym. Chem., 2005, 43, 1757–1766 CrossRef CAS .
- K. Wakabayashi, S. Kohama, S. Yamazaki and K. Kimura, Macromolecules, 2008, 41, 1168–1174 CrossRef CAS .
- M. Kihara, Y. Sakakiyama, S. Yamazaki and K. Kimura, Polymer, 2015, 66, 222–229 CrossRef CAS .
-
K. Kimura, in Polymer Science: A Comprehensive Reference, Elsevier, 2012, pp. 95–113 Search PubMed .
- H. R. Kricheldorf, M. G. Zolotukhin and J. Cárdenas, Macromol. Rapid Commun., 2012, 33, 1814–1832 CAS .
- T. Yokozawa and Y. Ohta, Polym. Sci., Ser. C, 2020, 62, 78–89 CrossRef .
- H. Gao, ACS Omega, 2021, 6, 4527–4533 CrossRef CAS PubMed .
-
T. Yokozawa and Y. Ohta, in Macromolecular Engineering, ed. N. Hadjichristidis, Y. Gnanou, K. Matyjaszewski and M. Muthukumar, Wiley, 1st edn, 2022, pp. 1–71 Search PubMed .
- T. Higashihara, J. Synth. Org. Chem., Jpn., 2023, 81, 574–581 CrossRef CAS .
-
T. Yokozawa and Y. Ohta, in Macromolecular Engineering, ed. N. Hadjichristidis, Y. Gnanou, K. Matyjaszewski and M. Muthukumar, Wiley, 1st edn, 2022, pp. 1–71 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*