
Aromatic vs. aliphatic linkers: impact on dye loading and stability in oligoglycerol-derived dendronized polymersomes†
Raj Kumar
Roy
a,
Trisha
Samanta
a,
Supriyo
Saha
a,
Aparna
Ramesh
b,
Naznin Ara
Begum
 a,
Goutam
Ghosh
b and
Pradip
Dey
*a
a,
Goutam
Ghosh
b and
Pradip
Dey
*a
aDepartment of Chemistry, Siksha Bhavana, Visva-Bharati (A Central University), Santiniketan, 731235 India. E-mail: pradip.dey@visva-bharati.ac.in
bCentre for Nano and Soft Matter Sciences (CeNS), Arkavathi, Survey No. 7, Shivanapura, Dasanapura Hobli, Bengaluru 562162, Karnataka, India
First published on 5th November 2024
Abstract
Nanocarriers protect the payload from degradation and enable specific targeting of the diseased tissue, thus reducing systemic toxicity. So, it is crucial and desirable to design a nanocarrier with specific nano-architectures that possess those essential characteristics. Considering the above aspects, we report the one-pot synthesis and self-assembly of oligoglycerol-based amphiphilic dendronized polythiourethanes consisting of an aliphatic or aromatic linker. During the ring opening of cyclodithiocarbonate, the generated thiols were utilized to conjugate first or zeroth-generation oligoglycerol dendrons (containing four or two hydroxyl groups) in one pot. Among them, the aromatic linker containing polymer ARM-PTU-G1-OH had a higher encapsulation ability for hydrophobic dyes (pyrene and Nile red) than the other aliphatic linker containing polymer ALP-PTU-G1-OH. Both the polymers had hydrodynamic diameters of 167 nm with PDI = 0.314–0.326. In addition, AFM results showed that ALP-PTU-G1-OH and ARM-PTU-G1-OH formed spherical aggregates with diameters of 136 ± 28 nm and 161 ± 35 nm, respectively. The formation of polymersomes was probed by encapsulating the hydrophilic dye calcein. Both the polymers were able to encapsulate calcein. Among them, the aromatic linker-containing polymer had 31% encapsulation efficiency, whereas the aliphatic linker-containing polymer had 14% encapsulation efficiency. To understand the stability of the polymersomes, a FRET study was performed by encapsulating DiO and DiI dyes individually in both the polymersomes, and then mixing and studying the evolution of FRET with time. The results showed that the mechanism of dye exchange was different for both polymers, and the aromatic linker containing polymersome had better stability during dye exchange. A few parameters were calculated by fitting the change in donor emission intensity with time, and it showed that in ARM-PTU-G1-OH dye exchange occurred via a slow merging and splitting mechanism. In contrast, in ALP-PTU-G1-OH, a fast expulsion and insertion mechanism was mainly operative.
Introduction
Introducing nanotechnology for cancer therapy may improve the efficiency of the current anti-cancer drugs and quality of life in cancer patients during treatment. Thus, polymer therapeutics have emerged as a challenging and growing field.1–3 The central concept of using nanocarriers for drug delivery (such as low molecular weight and therapeutic proteins) has evolved in the last few decades. The advantages are the following: (i) the vehicle protects the payload from degradation in vivo; (ii) specific targeting to the diseased tissue can be achieved using the Enhanced Permeability and Retention (EPR) effect; and thus (iii) the risk of systemic toxicity is reduced; and finally, (iv) the drug is released and the carrier is eliminated from the body.4,5 Research on the encapsulation of hydrophobic molecules6–8 has demonstrated that appropriate transporter architectures with improved pharmacokinetic profiles and precise biodistribution are needed to optimize drug delivery processes to target specific diseases.9,10 More significant, precisely defined, and biocompatible molecular objects are therefore being developed as potential drug delivery devices.11 One method for creating these kinds of macromolecular nanostructures is to use polymers as multifunctional polydisperse cores to which dendrons are attached at each repeating unit through pendant functional groups; these polymers are known as “dendronized polymers”.12,13The micellar and liposomal structures are sensitive to dilution as their stability depends on the critical aggregation concentration (CAC) for self-assembly. Leaching of the encapsulated drug in the bloodstream is a major inherited problem in micellar systems due to stability and infinite dilution during intravenous application. In this regard, amphiphilic polymers are more advantageous than small molecule-based amphiphiles due to their low CAC and high stability in aqueous solutions.
Polythiourethane (PTU) chemistry is an exciting choice for designing nanocarriers for the following reasons.14–16 The polymerization is fast, and atom-economical, and can be done at room temperature. Free thiols generated during the polyaddition of bifunctional five-membered cyclodithiocarbonates (DTCs) with diamines could be applied for coupling the hydrophilic units, etc., compared to polyurethanes (Fig. 1a). PTUs are synthesized using a greener approach compared to traditional polyurethane synthesis, where isocyanates are used, which are toxic due to the presence of phosgene.16,17 The hydrophobic PTU will have H-bonding capability, forming an assembly instead of hydrophobicity-driven collapse of the polymeric chains, ensuring stability by orientating hydrophilic parts.18
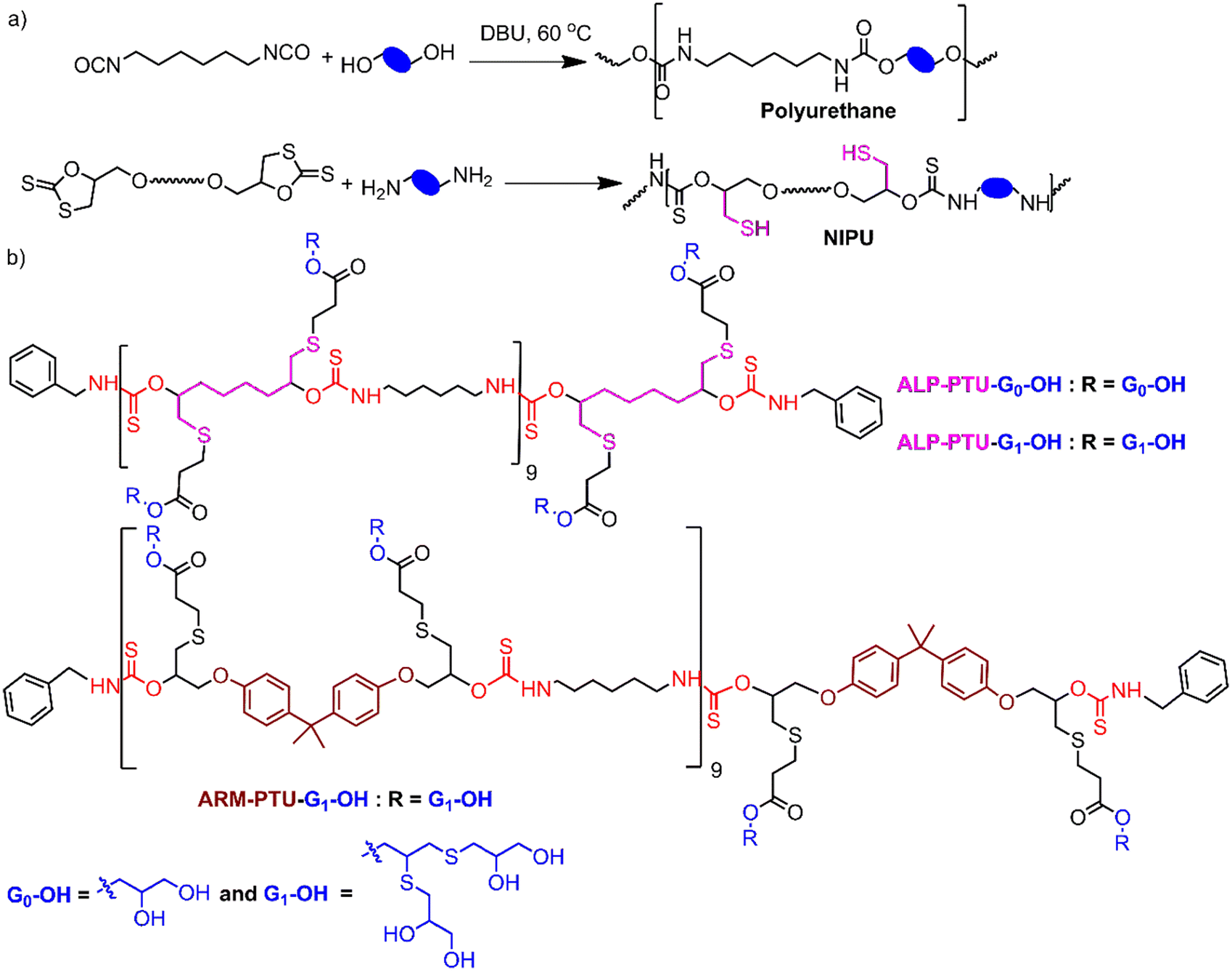 | ||
| Fig. 1 (a) Synthesis of polyurethane and polythiourethanes (PTU); (b) structure of the polymers. | ||
The study presents a green method for synthesizing oligoglycerol-based dendronized amphiphilic PTU in one pot, hypothesizing that these dendrons can enhance the general stability of supramolecular structures.19 Oligoglycerol dendrons, known for their biocompatibility and lack of immunogenicity, antigenicity, or toxicity, may offer superior performance over traditional monomolecular amphiphiles.20–22 Here, we have addressed the effect of the linker's flexibility on the stability of the formed aggregates due to the folding of the PTU backbone by studying the evolution of the acceptor dye's fluorescence which encapsulates the donor and acceptor dye individually in the formed aggregates.
Results and discussion
Polymer synthesis and characterization
The oligoglycerol-based dendrons and PTU backbone act as hydrophilic and hydrophobic moieties, respectively (Fig. 1b). The polymer was prepared by polymerizing bifunctional DTCs and hexyl diamine following the modified Carothers equation. To study the effect of the linker, two different DTC-based monomers (M1 and M2) were prepared by reacting carbon disulphide with 1,2,7,8-diepoxyoctane and 2,2-bis[4-(glycidyloxy)phenyl]propane, respectively, in the presence of LiBr (Schemes S1 and S2†). Oligoglycerol-based dendrons were introduced to the PTU backbone by quenching the thiols formed during polymerization due to the opening of the DTC ring in the presence of dendron-acrylate (Fig. 2). M1 and M2 were characterized by 1H NMR, 13C NMR, FTIR, and HRMS (Fig. S1–S8†). Both zeroth and first-generation dendrons (having two and four hydroxyl groups, respectively) were functionalized with the acrylate group by EDC coupling of the corresponding solketal (HO-G0-P) and the first generation of oligoglycerol dendrons (HO-G1-P) (Schemes S4 and S5†). HO-G1-P was synthesized by grafting thioglycerol on propargylic alcohol by the thiol–ene reaction using the thermal initiator AIBN and further protecting the diol in situ using 2,2-dimethoxypropane (Scheme S3†) and characterized by 1H NMR, 13C NMR, and HRMS (Fig. S9–S11†).23 Similarly, Acr-G0-P and Acr-G1-P were characterized by 1H NMR and HRMS (Fig. S12–S15†). All the polymers (ARM-PTU-G1-P, ALP-PTU-G0-P, and ALP-PTU-G1-P) were synthesized by polycondensation between M1 or M2 (AA) and the hexyl diamine derivative (HDA, BB) in the presence of benzylamine (MFI, B*) which acted as the chain stopper (Fig. 2, Schemes S6–S9†). AA, BB, and B* were mixed in a particular ratio so that 2NAA = 2NBB + NB*, wherein NAA, NBB, and NB* are the mole fractions of M1 (or M2), HDA, and the chain stopper, respectively (Table S1†).24 From the end group analysis, by comparing the intensity at 7.26 ppm of the phenyl ring proton (benzyl group) and the corresponding peaks at 4.0–4.5 ppm (oligoglycerol backbone protons), the number of repeating units (DPn) was found to be 10 (Fig. S16–S18†). Fig. 3 shows the incorporation of dendrons in the PTU backbone. Furthermore, the polymers were deprotected using TFA in dry DCM (Fig. 2).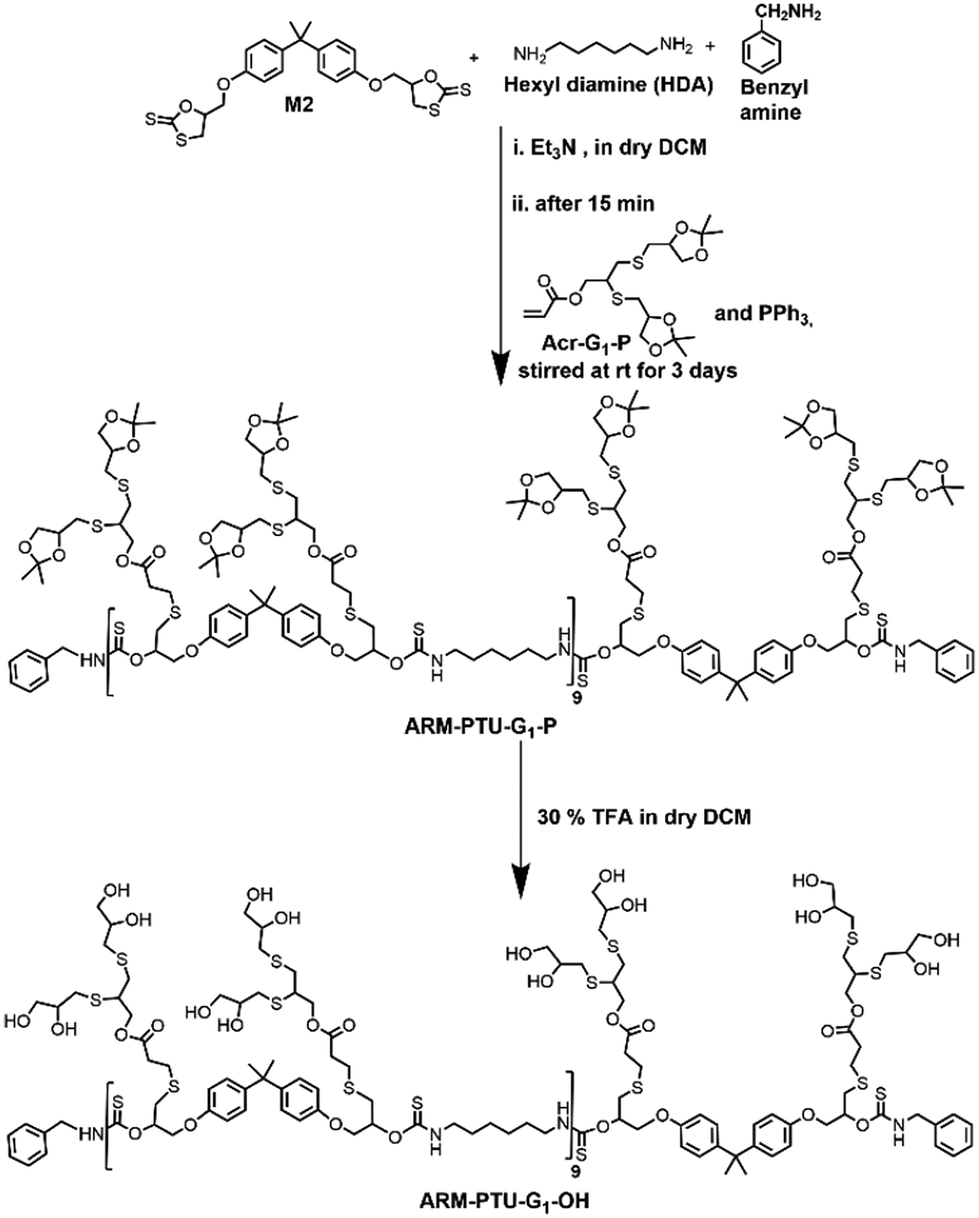 | ||
| Fig. 2 Schematic showing the synthesis of ARM-PTU-G1-OH. | ||
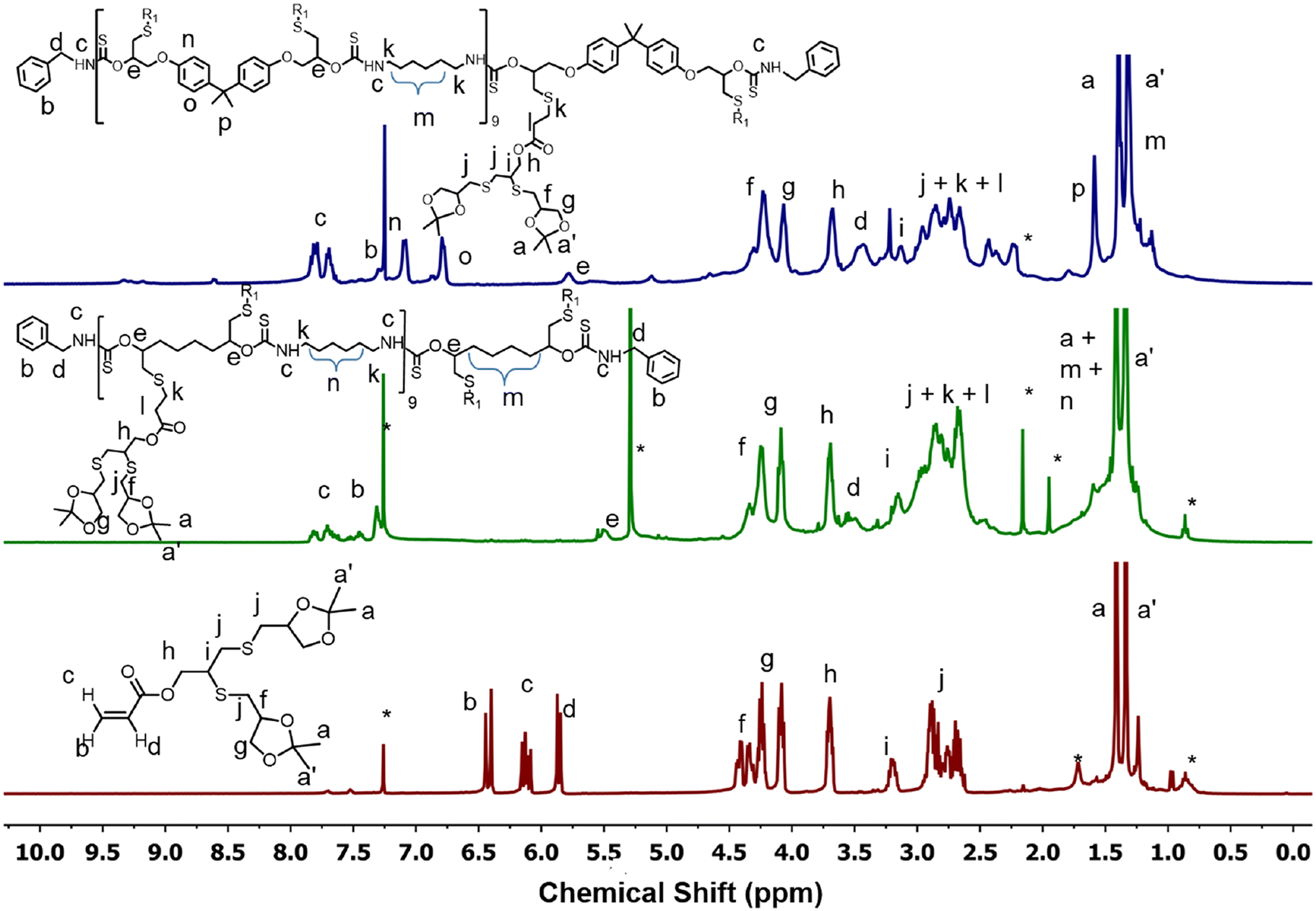 | ||
| Fig. 3 Comparison of 1H NMR spectra of Acr-G1-P, ALP-PTU-G1-P and ARM-PTU-G1-P. * indicates solvent impurities. | ||
The disappearance of the peak at 1.3 ppm confirms the successful deprotection of acetal groups (Fig. S19–S21†). The polymers were characterized by FTIR (Fig. 4 and S22†) and SEC (Fig. S23†). In the FTIR spectra of both ALP-PTU-G1-OH and ARM-PTU-G1-OH, the prominent peaks at 2929, 1680, and 1525 cm−1 are assigned to the C–H stretching and N–H bending of the thiourethane linkage with the disappearance of the FTIR band at 1182 cm−1 of M1 and M2 (Fig. 4 and S22†), respectively, which confirm the successful synthesis of PTU. Mn was found to be 2144 (Đ = 1.12) and 2237 g mol−1 (Đ = 1.17) for ALP-PTU-G1-OH and ARM-PTU-G1-OH, respectively (Table S1†). The molecular weight obtained by SEC did not match the molecular weight obtained from the end group analysis (Fig. S23†), as SEC provides the relative molecular weight with respect to a standard.
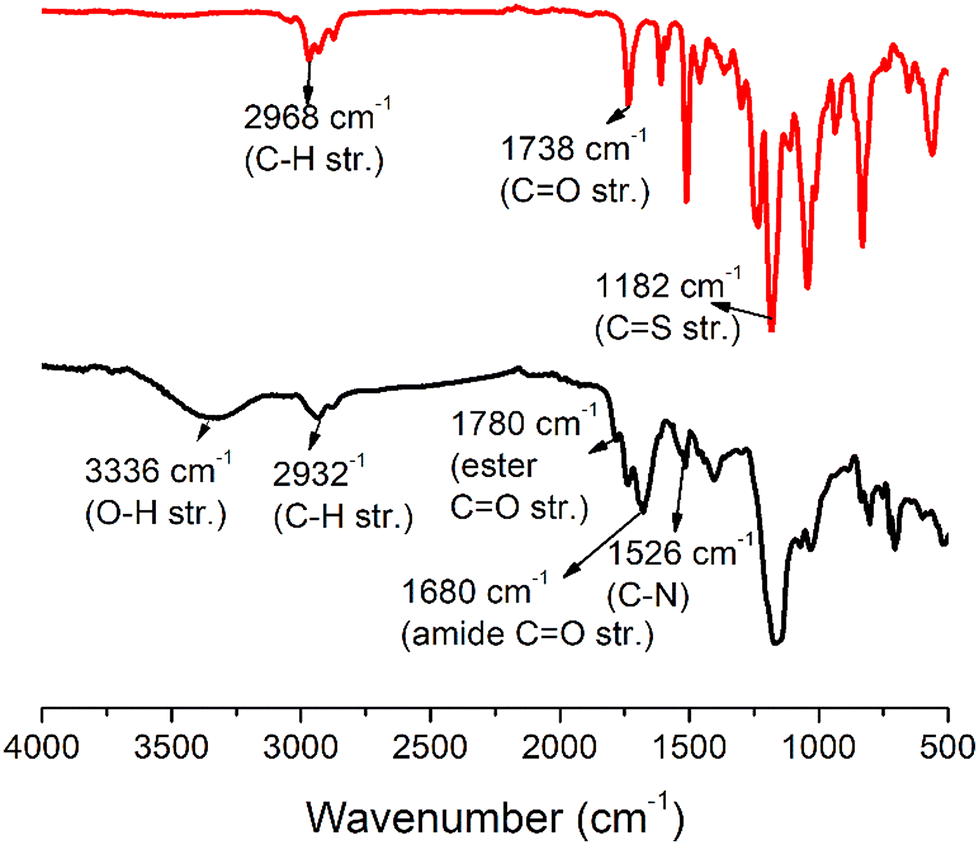 | ||
| Fig. 4 Comparison of the FTIR spectrum of M2 (black line, top) and ARM-PTU-G1-OH (red line, bottom). | ||
Aqueous self-assembly
Next, the critical aggregation concentrations (CACs) of the synthesized polymers were determined by encapsulation of Nile red (Fig. 5). ALP-PTU-G1-OH (Fig. 5a) and ARM-PTU-G1-OH (Fig. 5b) had CACs around 66 and 56 μM, respectively. DLS of the aqueous solution of ALP-PTU-G1-OH and ARM-PTU-G1-OH (c = 1 mg mL−1) showed a single sharp peak (Fig. 6) with an average diameter of 167.7 nm (PDI = 0.326) and 167.9 nm (PDI = 0.314), suggesting the formation of aggregates (autocorrelation functions of both measurements are given in Fig. S24†). AFM (Fig. 7) revealed the presence of spherical aggregates with diameters in the range of 136 ± 28 nm (n = 20) (Fig. 7b) and 161 ± 35 nm (n = 35) (Fig. 7d) for ALP-PTU-G1-OH and ARM-PTU-G1-OH, respectively. As AFM was done under dry conditions, whereas DLS provides the hydrodynamic diameter, it can be concluded that the AFM result roughly corroborates with that estimated from DLS.25 Moreover, as demonstrated in the height profiles (inset, Fig. 7a), the height of ALP-PTU-G1-OH was in the range of 70–120 nm, whereas for ARM-PTU-G1-OH the height was 18–23 nm (inset, Fig. 7c). Even though both had similar hydrodynamic diameters, we observed different diameters that could be correlated with the height data. It can be concluded that ARM-PTU-G1-OH was flatter on the mica surface, whereas ALP-PTU-G1-OH was more compact, which is why the difference in the diameter was observed.25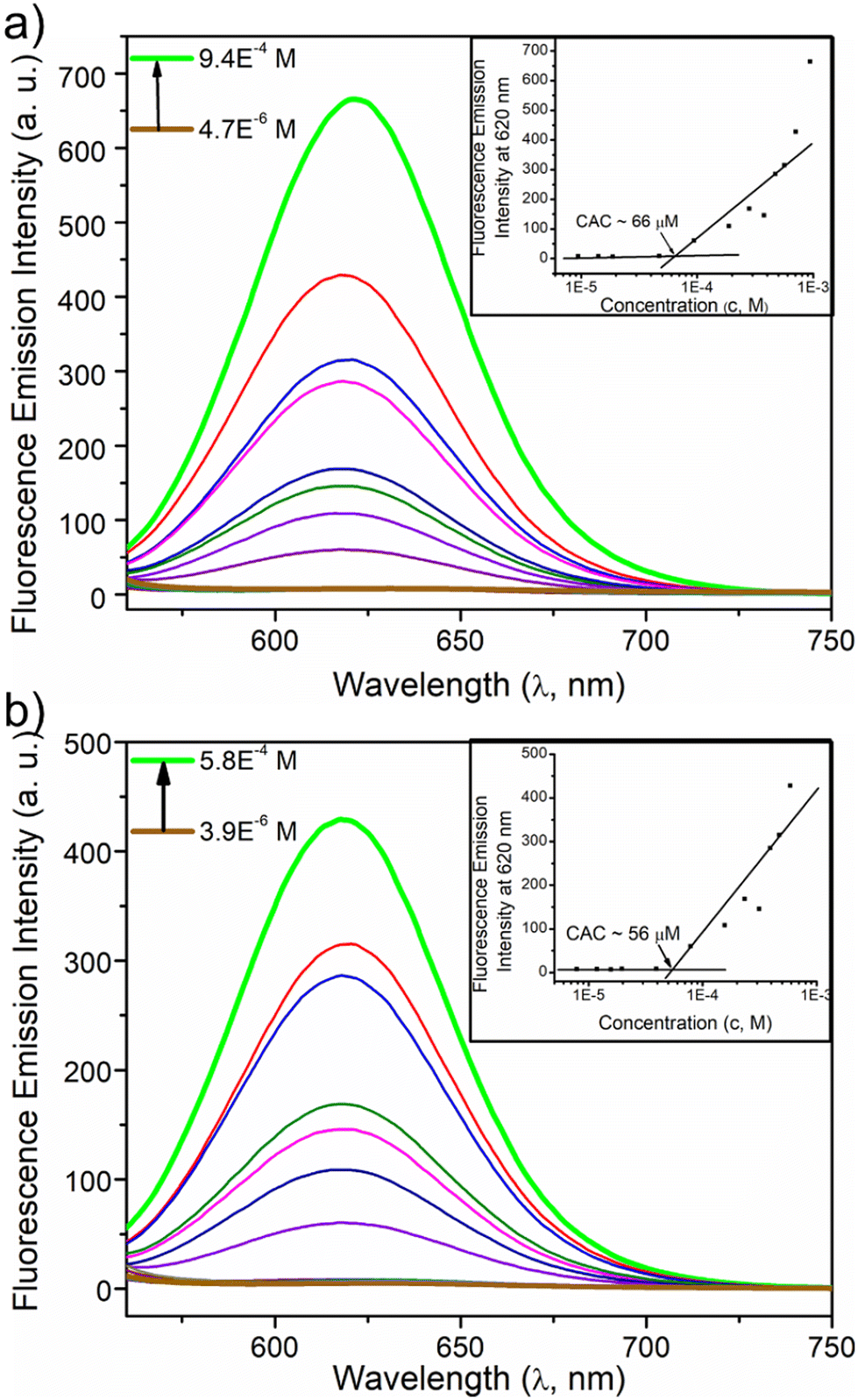 | ||
| Fig. 5 Emission spectra of Nile red encapsulated in the aqueous solution of (a) ALP-PTU-G1-OH and (b) ARM-PTU-G1-OH at varying concentrations. Inset showing the variation of I620 in Nile red emission spectra as a function of ALP-PTU-G1-OH and ARM-PTU-G1-OH concentrations. The Nile red concentration was kept constant (c = 10.0 μM). | ||
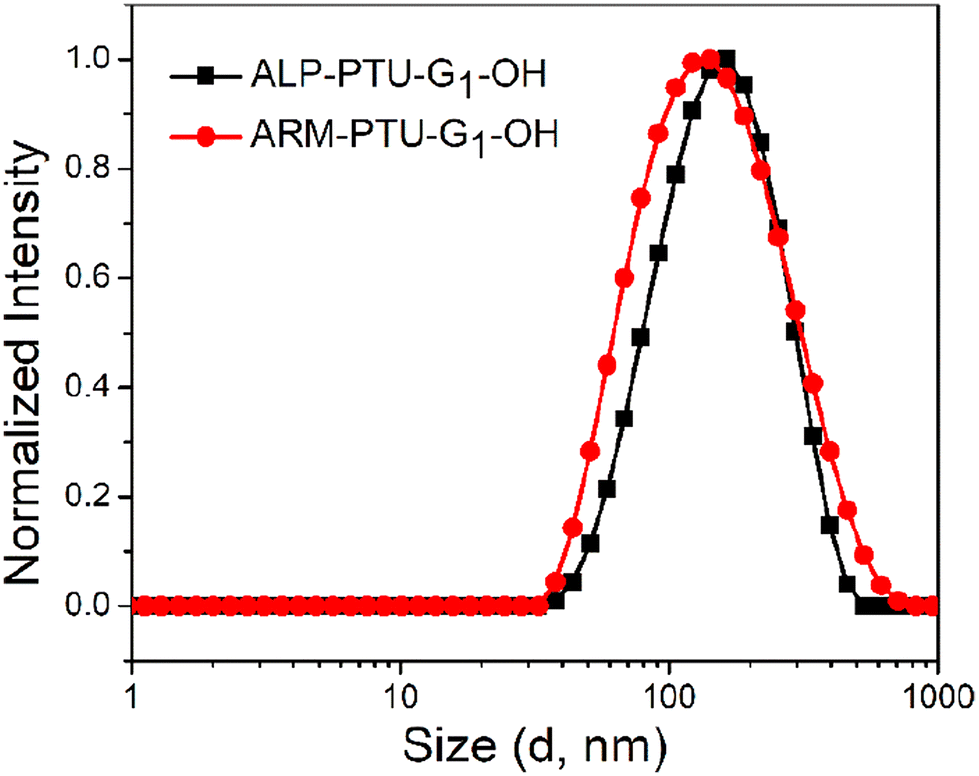 | ||
| Fig. 6 DLS comparison of ALP-PTU-G1-OH and ARM-PTU-G1-OH (c = 1 mg ml−1). | ||
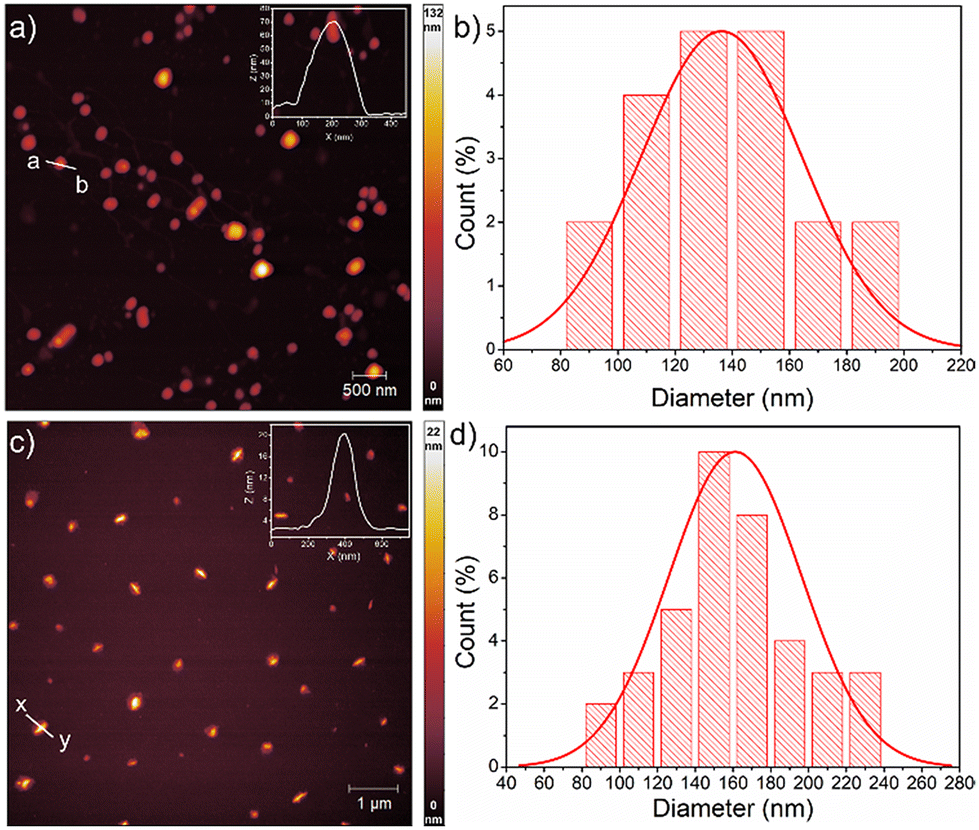 | ||
| Fig. 7 AFM image of (a) ALP-PTU-G1-OH and (c) ARM-PTU-G1-OH in water (c = 1 mg mL−1). Height profile shown in the inset is for (a, b) and (x, y). (b) and (d) Size distribution obtained analyzing the AFM images of ALP-PTU-G1-OH and ARM-PTU-G1-OH, respectively, using image J software. | ||
Hydrophilic dye encapsulation
The hydrophilic container property of ALP-PTU-G1-OH and ARM-PTU-G1-OH aggregates was demonstrated by encapsulating the hydrophilic fluorescent dye calcein (Fig. 8). Both the polymers could encapsulate the hydrophilic dye and showed absorption bands in the 425–550 nm range (Fig. 8a and c). The encapsulation efficiency of calcein was estimated to be 14% and 31%, respectively, for ALP-PTU-G1-OH and ARM-PTU-G1-OH. Absorption-matched fluorescence spectra (Fig. 8b and d) of the free and entrapped dye indicated considerable quenching, indicating its existence in a constrained environment.26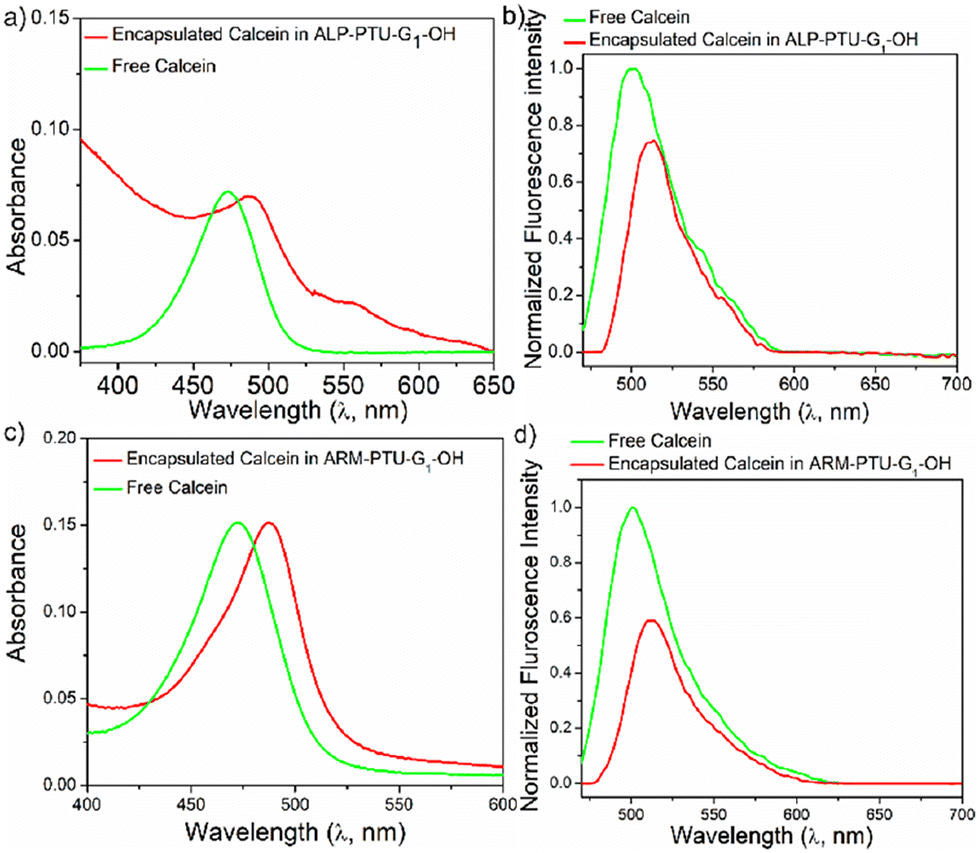 | ||
| Fig. 8 (a) and (c) UV/vis, and (b) and (d) concentration matched fluorescence spectra of calcein encapsulated aqueous aggregate of ALP-PTU-G1-OH and ARM-PTU-G1-OH (c = 1 mg mL−1) and the free dye. In the fluorescence spectra, the band intensity was normalized with respect to the absorption intensity of the respective sample in water. | ||
Hydrophobic dye encapsulation
Though no significant difference in CACs was observed for both the polymers, we investigated the container property by encapsulating hydrophobic dyes such as Nile red and pyrene. ALP-PTU-G0-OH did not show any ability to encapsulate any hydrophobic dye like Nile red (Fig. 9b) or pyrene (Fig. 9a). So, we focused only on the other two polymers. ALP-PTU-G1-OH and ARM-PTU-G1-OH could encapsulate both dyes (Table S2†). ARM-PTU-G1-OH showed better pyrene loading (3.6 mg of pyrene per g of polymer) than ALP-PTU-G1-OH (1.32 mg of pyrene per g of polymer) (Fig. 9a).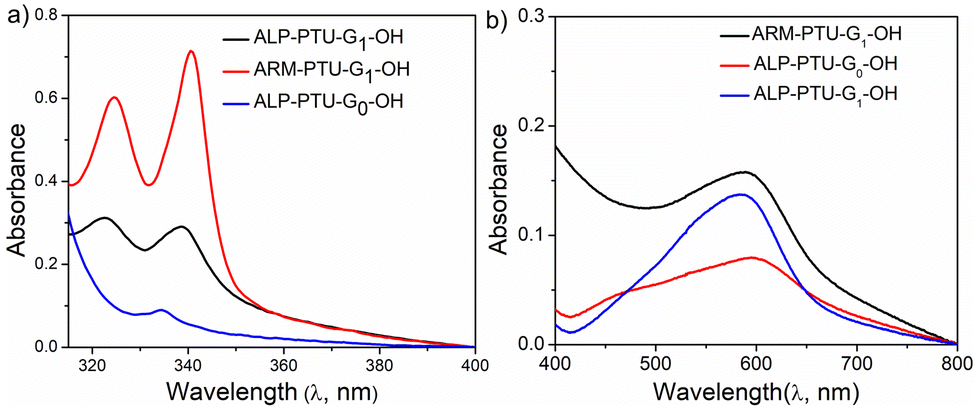 | ||
| Fig. 9 UV/Vis spectra of (a) pyrene and (b) Nile red encapsulated in the aqueous solution of ALP-PTU-G0-OH, ALP-PTU-G1-OH, ARM-PTU-G1-OH. Pyrene and Nile red concentration was kept constant 1.0 μM and 10.0 μM, respectively. | ||
FRET studies
Fluorescence resonance energy transfer (FRET) experiments have been utilized to probe the exchange dynamics and indirectly understand the noncovalent encapsulation stability.27 Two lipophilic dyes, namely, DiO and DiI were used in the FRET studies (Fig. S25†). They were separately encapsulated in polymersomes of ALP-PTU-G1-OH and ARM-PTU-G1-OH in water. Then, the solutions were mixed, and the encapsulation stability was checked by fluorescence spectroscopy, and correlated with the chain exchange dynamics of the polymersome.28 As expected, the dyes were segregated in separate polymersomes, exhibiting initial low FRET for both aggregates with a weak acceptor emission (λmax = 577 nm) and a significant donor emission (λmax = 510 nm) (Fig. 10).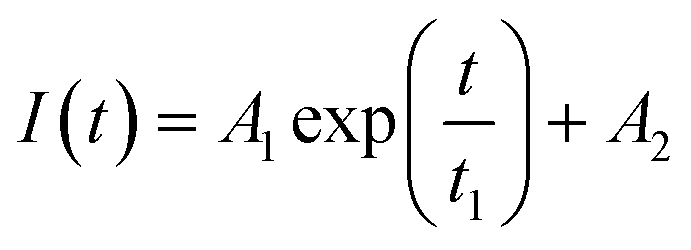 | (1) |
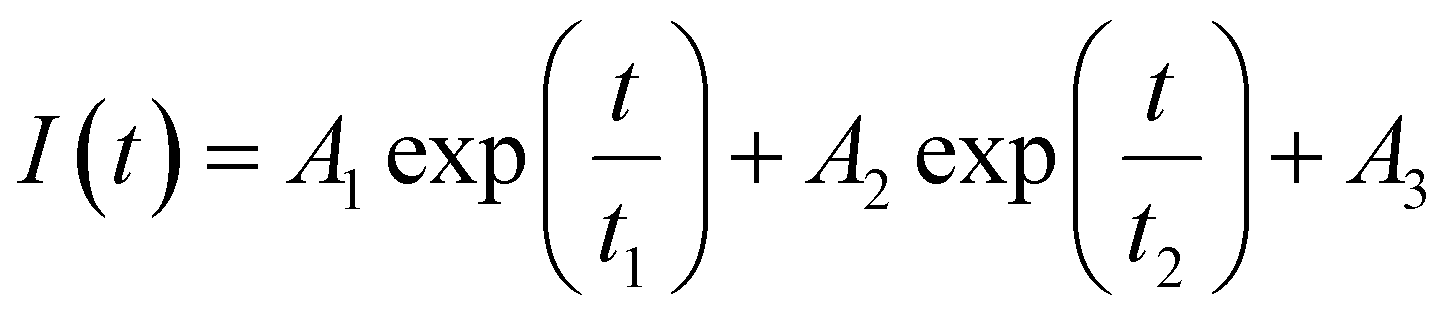 | (2) |
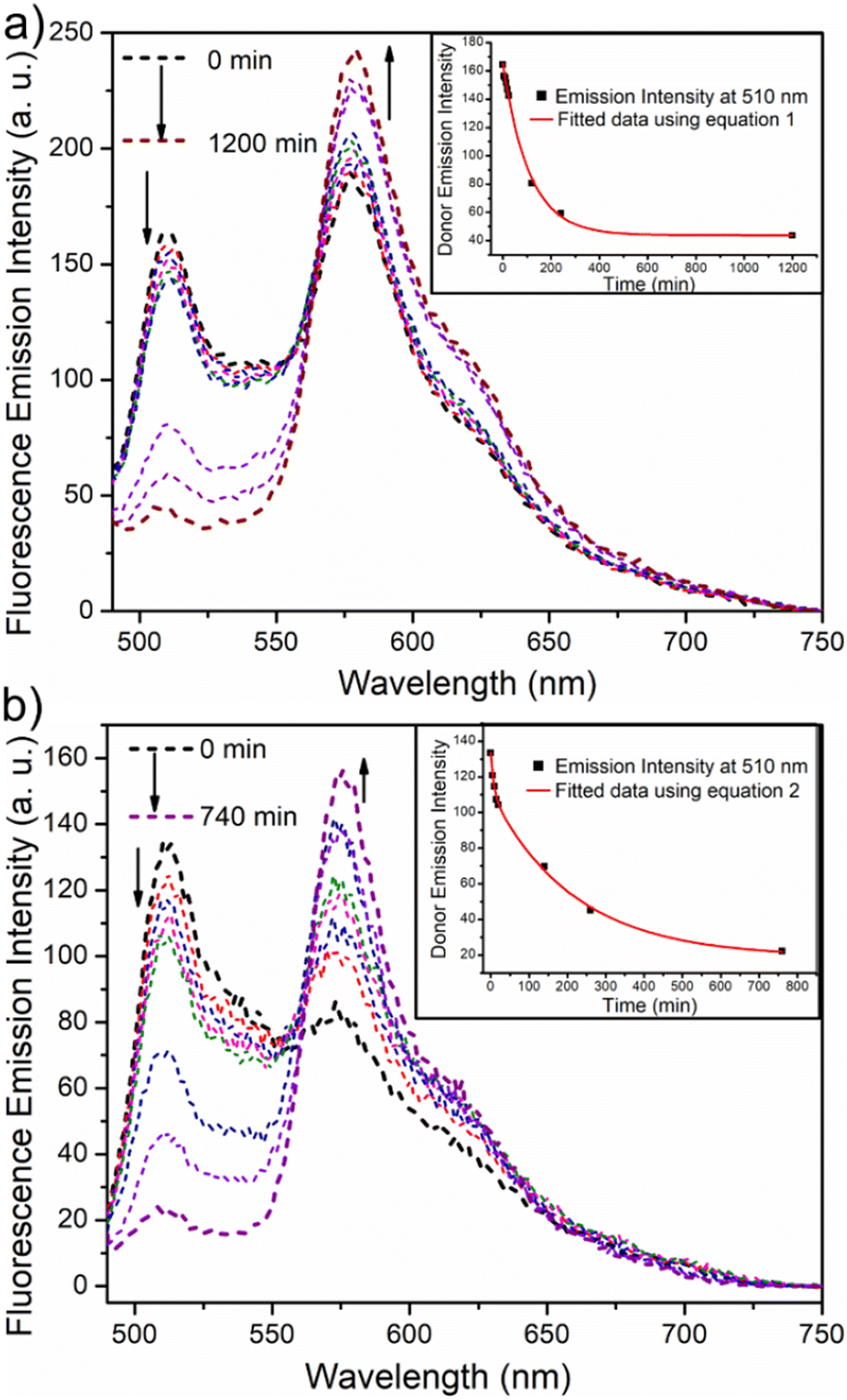 | ||
| Fig. 10 Emission spectra (λex = 470 nm) of a mixture of DiO-encapsulated (0.5 mM) and DiI-encapsulated (0.5 mM) (a) ALP-PTU-G1-OH (1 mg mL−1) and (b) ARM-PTU-G1-OH (1 mg mL−1) in water at different time intervals. The direction of the spectrum shift over time is marked by the arrows. The change in donor emission intensity (λem = 510 nm) over time is displayed in the inset. The red line is the fitted data using eqn (1) (inset a) and (2) (inset b). | ||
The donor emission decreased over time (indicated by the arrow) due to the exchange of two dyes between polymersomes, while the acceptor emission increased simultaneously, resulting from FRET. The time-dependent change in the donor emission intensity for ALP-PTU-G1-OH can be fitted satisfactorily using eqn (1) (exponential decay) with an adj. R-square value of 0.9968 (inset, Fig. 10a). On the other hand, for ARM-PTU-G1-OH, the donor emission intensity cannot be reasonably fitted using eqn (1) (Fig. S26†), suggesting that several mechanisms are involved during the exchange.29 Alternatively, the donor fluorescence emission intensity of ARM-PTU-G1-OH could be fitted using eqn (2) (biexponential decay) with an adj. R-square value of 0.99754 (inset, Fig. 10b), where t1 and t2 represent the relaxation times for the faster (pathway 1) and slower (pathway 2) exchange processes, respectively (Fig. 11b).30 A1 and A2 represent the preexponential factors of the two pathways. A3 is a constant in this equation. A summary of all the chain exchange parameters in this fitting for both aggregates is presented in Table 1. Pathway 1 is primarily responsible for the lifetime t1 = 121 min (with about 100% contribution) in ALP-PTU-G1-OH, which is explained by the chain expulsion or insertion process (Fig. 11b). In contrast, pathway 2, which has a significantly longer lifetime (t2 = 219 min) and is dominant (about 81%) for ARM-PTU-G1-OH, is thought to be the consequence of polymersome combination or splitting. Like block copolymer micelles, the faster exchange pathway's lifespan (t1 = 8 min, ∼19%) is attributed to the chain expulsion/insertion mechanism (pathway 1), and compared to the polymersome generated by ALP-PTU-G1-OH, the average lifetime 〈tavg〉 increased for ARM-PTU-G1-OH (177 min). As a result, it is evident that for ARM-PTU-G1-OH, the overall exchange dynamics decreased, and the expulsion/insertion mechanism became less controlled while the polymersome's merging/splitting process became more active.
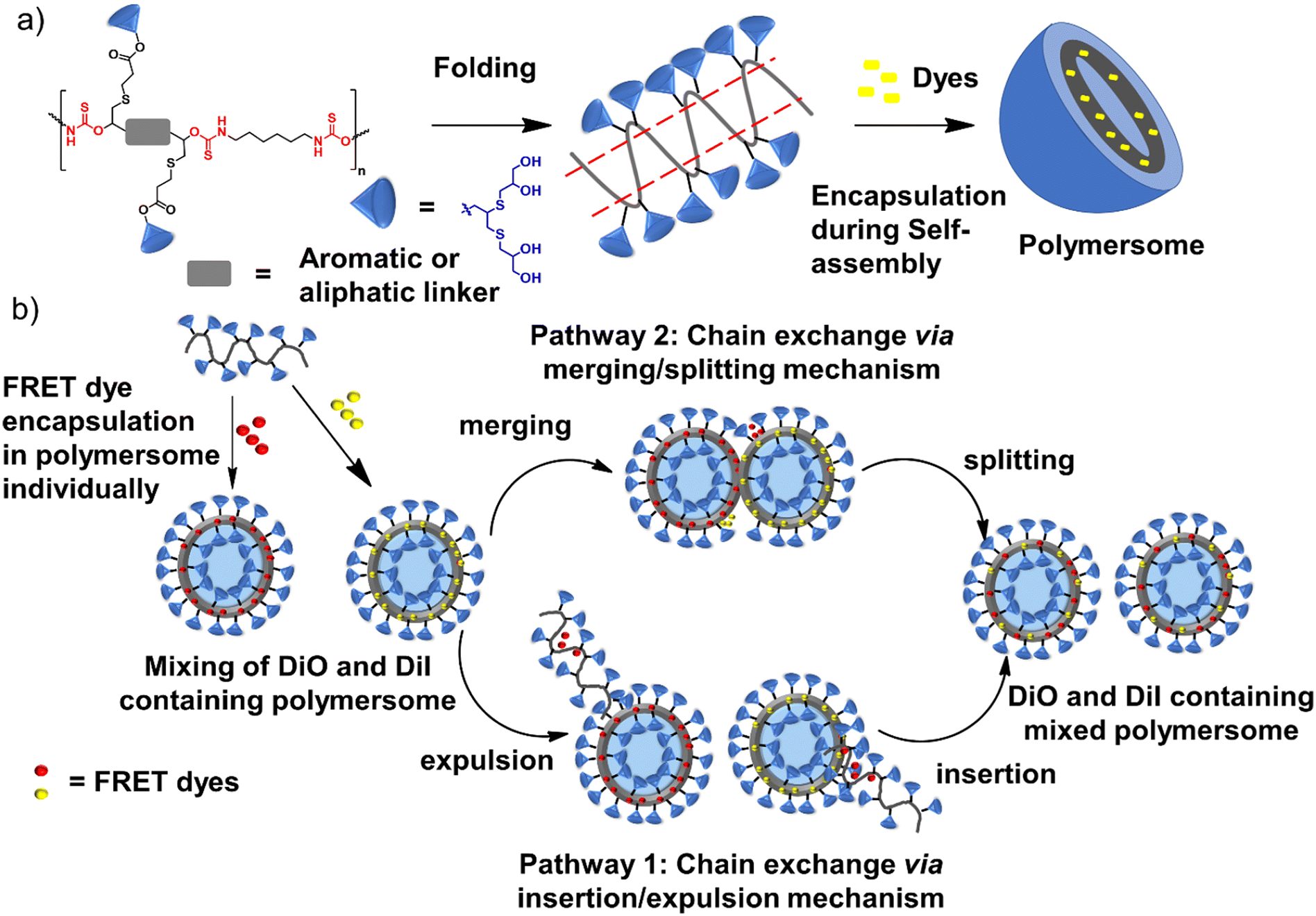 | ||
| Fig. 11 (a) Schematic description of polythiourethane chain folding induced polymersome formation and dye encapsulation in water, (b) possible chain exchange mechanism in polymersome formed by ALP-PTU-G1-OH (pathway 1, major contribution) and ARM-PTU-G1-OH (pathway 2, major contribution). | ||
| Polymers | t 1 (min) | t 2 (min) | f 1 | f 2 | 〈tavg.〉 (min) | Adj. R-square |
|---|---|---|---|---|---|---|
| ALP-PTU-G1-OH | 121.40 | — | 1.0 | — | NA | 0.9968 |
| ARM-PTU-G1-OH | 8 | 219 | 0.19 | 0.81 | 177.6 | 0.99754 |
To compare the observed guest stability of ALP-PTU-G1-OH and ARM-PTU-G1-OH, pyrene was encapsulated in ALP-PTU-G1-OH and ARM-PTU-G1-OH individually, and the UV/vis spectra of the aqueous pyrene-encapsulated solutions were recorded at different time points (Fig. S28†). Pyrene absorption decreased with time in both solutions, but the decrease in absorption was faster for ALP-PTU-G1-OH than for ARM-PTU-G1-OH. For ARM-PTU-G1-OH, more than 80% pyrene was released after 48 h, whereas for ALP-PTU-G1-OH, more than 80% pyrene was released in 4 h.
Experimental details
Chemicals
Solvents and reagents were purchased from commercial sources and purified following the reported procedure.31 1,2,7,8-Diepoxyoctane, propargylic alcohol, 2,2′-azobis(2-methylpropionitrile) (AIBN), 1-thioglycerol, and 2,2′-dimethoxypropane were purchased from Sigma Aldrich. p-Toluenesulfonic monohydrate (pTSA·H2O), 1,6-diaminohexane, lithium bromide, acrylic acid, triethylamine, and triphenylphosphine were purchased from Tokyo Chemical Industry (TCI). Carbon disulphide was purchased from Nice Chemicals Private Limited. 2,2-Bis(4-glycidyloxyphenyl) propane was purchased from BLD Pharmatech (India) Pvt. Ltd. Benzylamine, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimidehydrochloride (EDC·HCl) and 4-dimethyl amino pyridine (DMAP) were purchased from Sisco Research Pvt. Ltd. SnakeSkin™ Dialysis Tubing was bought from Thermo Fisher Scientific (Thermo Scientific™) with a molecular weight cut-off (MWCO) of 3.5 kDa.Characterization
Synthesis of ALP-G0-P
ALP-PTU-G0-P was synthesized following the procedure presented in Scheme S6.† Benzylamine (0.14 eq., 0.011 g, 0.099 mmol) and hexylamine (1.9 eq., 0.152 g, 1.3 mmol) were taken in a flat-bottomed glass tube (specially made) containing a small magnetic bead under N2 gas. To it, 0.3 mL of dry DCM and triethylamine (4.2 eq., 0.291 g, 2.88 mmol) were added and the mixture was stirred for 10 minutes at room temperature. The dithiocarbonate monomer (M1) (1 eq., 0.2 g, 0.68 moles) was taken in a glass vial and dissolved in a minimum volume of dry DCM. Then the resulting solution was added to the reaction mixture and stirred for 15 minutes. Then PPh3 (0.2 eq., 0.035 g, 0.135 mmol) and Acr-G0-P (2.5 eq., 0.27 g, 1.7 mmol) were added to the above mixture by dissolving in dry DCM (0.5 mL). After 3 days, the polymer was precipitated in cold hexane three times by dissolving in DCM. The precipitate was collected via centrifugation. Upon drying under high vacuum, ALP-PTU-G0-P (0.301 g, yield = 65%) was obtained as a brown liquid.1H NMR (400 MHz, CDCl3): δ (ppm) = 7.75–7.39 (m, 45H), 7.23–7.27 (d, 10H), 5.51–5.30 (m, 2H), 4.44–3.75 (m, 56H), 3.76–3.15 (m, 40H), 3.14–2.35 (m, 66H), 1.60–1.23 (m, 122H).
Synthesis of ALP-G1-P
ALP-PTU-G1-P was synthesized (Scheme S7†) using the same procedure as described above, except that Acr-G1-P (2.5 eq., 0.690 g, 1.7 mmol) was used as the quenching agent instead of Acr-G0-P. The yield obtained was 65% (300 mg). The following reagents were used: benzylamine (0.14 eq., 0.01 g, 0.099 mmol), 1,6-diaminohexane (1.9 eq., 0.152 g, 1.31 mmol), triethylamine (4.2 eq., 0.291 g, 2.88 mmol), monomer (M1) (1 eq., 0.2 g, 0.68 moles) and PPh3 (0.2 eq., 0.035 g, 0.135 moles).1H NMR (400 MHz, CDCl3): δ (ppm) = 7.84–7.44 (m, 12H), 7.31–7.28 (dd, 10H), 5.55–5.51 (d, 6H), 4.34–3.78 (m, 154H), 3.35–2.25 (m, 418H), 1.30–1.25 (m, 302H).
Synthesis of ARM-PTU-G1-P
ARM-PTU-G1-P was synthesized (Fig. 2) using the same procedure as described above, except that M2 and Acr-G1-P (2.5 eq., 0.690 g, 1.7 mmol) were used as the quenching agent instead of M1 and Acr-G0-P. The yield obtained was 65% (0.501 g). The following reagents were used: benzylamine (0.14 eq., 0.011 g, 0.099 mmol), 1,6-diaminohexane (1.9 eq., 0.152 g, 1.31 mmol), PPh3 (0.2 eq., 0.035 g, 0.135 mmol), and Acr-G1-P (2.5 eq., 0.690 g, 1.7 mmol).1H NMR (400 MHz, CDCl3): δ (ppm) = 7.84–7.29 (m, 55H), 7.12–7.08 (d, 25H), 6.81–6.75 (d, H), 5.72–5.70 (m, 13H), 4.48–3.91 (m, 157H), 3.91–3.49 (m, 105H), 3.46–2.90 (m, 269H), 1.75–1.45 (s, 97H), 1.35–1.15 (m, 210H).
Synthesis of ALP-G0-OH
ALP-PTU-G0-OH was synthesized following the procedure presented in Scheme S8.† ALP-PTU-G0-P (0.150 g) was taken in a 50 mL round bottom flask equipped with a magnetic bead. Then 2 ml of dry DCM was added under N2 gas. To it, 30% CF3COOH in dry DCM was added dropwise using a syringe at 0 °C and stirred for 2.5 hours. Then, the solvent was evaporated using a rotavapor. The crude product was dissolved in a minimum amount of DCM. Then, the solution was precipitated in cold diethyl ether thrice. The precipitate was collected via centrifugation. The crude product was dried under vacuum, yielding ALP-PTU-G0-OH (0.144 g, yield 98%).1H NMR (400 MHz, CDCl3): δ (ppm) = 7.77–7.64 (m, 40H), 7.49–7.45 (d, 10H), 5.67–5.30 (m, 7H), 4.91–3.77 (m, 58H), 2.90–2.25 (m, 62H), 1.27–1.23 (m, 51H).
FTIR (cm−1): 3346, 2923, 1731, 1681, 1529, 1398, 1165, 1074, 1024, 811, 751, 690, 508 cm−1.
Synthesis of ALP-PTU-G1-OH
ALP-PTU-G1-OH was synthesized following the procedure presented in Scheme S9.† ALP-PTU-G1-P was deprotected as described above (yield 98%, 195 mg).1H NMR (400 MHz, CDCl3): δ (ppm) = 7.837–7.41 (m, 45H), 5.43–5.01 (m, 36H), 4.49–3.80 (m, 139H), 3.25–2.35 (m, 65H), 1.45–1.25 (m, 35H).
FTIR (cm−1): 3334, 2925, 1660, 1524, 1775, 1680, 1402, 1150, 1035, 797, 687.
Synthesis of ARM-G1-OH
ARM-PTU-G1-OH was synthesized following the procedure presented in Fig. 2. ARM-PTU-G1-P was deprotected as described above (yield 96%, 240 mg).1H NMR (400 MHz, CDCl3): δ (ppm) = 7.85–7.38 (m, 45H), 7.14–7.10 (d, 15H), 6.76–6.74 (d, 17H), 5.28–5.26 (m, 25H), 4.80–4.01 (m, 93H), 3.91–3.51 (m, 145H), 3.46–2.47 (m, 110H), 1.75–1.45 (s, 38H), 1.40–1.10 (m, 23H).
FTIR (cm−1): 3334, 2972, 2933, 2883, 1735, 1775, 1680, 1524, 1173, 1035, 804, 700, 515.
CAC determination and Nile red encapsulation
30 μL of Nile red stock solution in THF (c = 10–3 M) was taken in different vials. To these vials, different volumes of the polymer stock solution (10 mg mL−1) in THF were added. THF was removed by heating. 3 mL of double distilled water was added to each vial to obtain a series of solutions with constant Nile red concentration (10–5 M) and varying polymer concentration (from 3 μM to 1.0 mM). Each solution was sonicated for 20 minutes and kept at room temperature overnight. The spectrum was recorded on a PerkinElmer LS55 fluorescence spectrophotometer (λex = 536 nm, excitation slit = 7 nm, emission slit = 2.5 nm). The emission intensities at 620 nm were plotted against the concentration of the polymers. The inflection point in such a plot was considered as CAC (critical aggregation concentration). The UV absorption of encapsulated Nile red in different polymers of 1 mg mL−1 concentration was recorded on a Shimadzu UV/Vis spectrophotometer. The concentration of Nile red encapsulated in the polymer was determined using the calibration curve of Nile red in a 60% dioxane solution in water (v/v) (Fig. S27†).Pyrene encapsulation and release study
60 μL of the pyrene stock solution in THF (10−3 M) was taken in three different vials. To each vial, 4 mg mL−1 of three polymer solutions in THF was added. THF was removed by heating. The final volume was adjusted to 3 mL with double distilled water to obtain a final concentration of the solution of pyrene (10−6 M) and a polymer concentration of (1 mg mL−1). Each solution was sonicated for 20 minutes and kept at room temperature overnight. The amount of pyrene was calculated considering the molar absorptivity of 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1 at 338 nm wavelength.32 Similarly the UV/vis spectrum was recorded with the pyrene encapsulated solution at different time points.
500 M−1 cm−1 at 338 nm wavelength.32 Similarly the UV/vis spectrum was recorded with the pyrene encapsulated solution at different time points.
Calcein encapsulation
30 μL of the Calcein stock solution in MeOH (10−3 M) was taken in a vial. To each of these vials 10 mg mL−1 of polymer solution in THF was added. Then the THF was evaporated by heating, and 3 mL of double distilled water was added. The mixtures were sonicated for 20 minutes and subject to dialysis for 48 h to remove MeOH or any non-encapsulated dye. Then the final volume was adjusted to 3.0 mL by adding a measured amount of water to obtain the final concentration of the solution of calcein (10−5 M) and a polymer concentration of 1 mg mL−1.Sample preparation for AFM and DLS
In a vial, 300 μL (10 mg mL−1) of polymer solution in THF was taken, and 2.7 mL of double distilled water was added dropwise over an hour. Then the solution was dialyzed for 48 h changing the dialysate twice a day to remove THF. Then the solution inside the dialysis bag was collected and the volume was adjusted to 3 mL with double distilled water to make the concentration 1 mg mL−1. Before sample processing, the samples were filtered through 0.45 μm pore size nylon filters.Sample preparation for FRET studies
1.0 mM solution of the acceptor (A) dye DiI (1,10-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) and the FRET donor (D) dye DiO (3,3′-dioctadecyloxacarbocyanine perchlorate) were prepared in THF as stock solutions. The polymer solution in THF was combined with the measured volume of each dye solution. The solvent was then removed by air drying, and the measured volume of water was then added to each vial to bring the total polymer concentration to 1.0 mg mL−1 and the concentration of each dye to 10 μM. The resulting solutions were then sonicated for 15 min and kept overnight in the dark. Time-dependent emission spectra were recorded immediately after mixing donor and acceptor containing aggregated solutions and the emission of the mixed solution was monitored upon exciting the donor at 470 nm. Using Origin Pro 8.5 software, the donor fluorescence intensity decays were fitted to a single exponential (eqn (1)) and a biexponential equation (eqn (2)). The adj. R-square value, which was 0.99 for both fittings, indicated whether the fitting was acceptable (values near to 1 are considered to be the best match). From the pre-exponential components Ai, the observed fractional intensity contribution (fi) is approximated as fi = Ai/∑fiAi. The average lifetime 〈tavg.〉 was computed from fi using 〈tavg.〉 = ∑fiti. The constant A3 is a fixed constant.Conclusion
In this manuscript, we successfully demonstrated the synthesis of dendronized oligoglycerol-based amphiphilic polymers using step-growth polymerization of DTC and diamine. The produced thiols from the ring opening of DTC were used to conjugate the first and zeroth-generation oligoglycerol dendrons (which had two and four hydroxyl groups, respectively). Both the polymers could form an aggregate with CACs in the range of 56–66 μM with a hydrodynamic diameter of 167 nm with PDI = 0.314–0.326. Furthermore, according to AFM data, ARM-PTU-G1-OH and ALP-PTU-G1-OH generated spherical aggregates with diameters ranging from 161 ± 35 nm to 136 ± 28 nm, respectively. The aromatic linker containing polymer ARM-PTU-G1-OH showed a higher ability to encapsulate hydrophobic dyes (pyrene and Nile red) than the other aliphatic linker containing polymer ARM-PTU-G1-OH. The study investigated the formation of polymersomes by encapsulating the hydrophilic dye calcein, with the aromatic linker-containing polymer showing 31% encapsulation efficiency, whereas the aliphatic linker-containing polymer had 14% encapsulation efficiency. In addition, the aromatic linker in the PTU backbone could stabilize and slow down the exchange of encapsulated dyes better than the aliphatic linker, as demonstrated by the FRET experiment.Moreover, by fitting the change in donor emission intensity, the possible mechanism for exchange could be unravelled. We believe the stability of the polymersome formed by ARM-PTU-G1-OH is due to the rigidity of the aromatic linker compared to the aliphatic linker present in ALP-PTU-G1-OH. The aromatic linker helped in stabilizing the folded structure (Fig. 11a) and thus, increased the stability of the formed aggregates. This phenomenon could help in designing hierarchical stable nanostructures using the folded assembly driven by H-bonding. The methodology developed in this manuscript could be helpful in synthesizing ABA-type linear amphiphilic polymers where pendant groups could be introduced in the hydrophobic block by quenching with acrylate derivatives and further disulfide-mediated crosslinking to increase the stability of the amphiphilic polymers. In addition, polymer degradation could be introduced easily in this amphiphilic polymer by varying the diamine to release the encapsulated payload in the presence of stimuli like pH, reductive enviroment,33,34etc. Moreover, the presence of many hydroxy groups on the surface of the polymersome provides an attractive opportunity to decorate the surface with targeting groups such as cell-penetraing peptides/ligands in a multivalent fashion35,36 to target specific cells.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
PD thanks the Anusandhan National Research Foundation (ANRF) (previously SERB) and DST, India, for funding through the Start-up Research Grant (SRG) (File No. SRG/2022/000195) and the Department of Chemistry, Visva-Bharati for providing research facilities. GG acknowledges the Ramanujan Fellowship (File no. RJF/2022/000002), SERB (now ANRF), Government of India. RR, TS, and SS thank the UGC for Junior Research Fellowship. PD acknowledges Professor Suhrit Ghosh for allowing us to use GPC facilities at his laboratory and Dr. Atish Nag for running the GPC samples.References
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef.
- I. N. Kurniasih, J. Keilitz and R. Haag, Chem. Soc. Rev., 2015, 44, 4145–4164 RSC.
- J. Khandare, M. Calderón, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- B. Kang, T. Opatz, K. Landfester and F. R. Wurm, Chem. Soc. Rev., 2015, 44, 8301–8325 CAS.
- J. Li, J. Fan, R. Cao, Z. Zhang, J. Du and X. Peng, ACS Omega, 2018, 3, 7380–7387 CAS.
- Y. Chi, N. Li, J. Tu, Y. Zhang, X. Li and C. Shao, J. Lumin., 2010, 130, 512–515 CrossRef CAS.
- D. Kumar, S. A. Mohammad, A. Kumar, S. R. Mane and S. Banerjee, Polym. Chem., 2022, 13, 1960–1969 RSC.
- E. Fleige, R. Tyagi and R. Haag, Nanocarriers, 2013, 1, 1–9, DOI:10.2478/nanca-2013-0001.
- S. Gupta, R. Tyagi, V. S. Parmar, S. K. Sharma and R. Haag, Polymer, 2012, 53, 3053–3078 CrossRef CAS.
- F. Calik, A. Degirmenci, M. Eceoglu, A. Sanyal and R. Sanyal, Bioconjugate Chem., 2019, 30, 1087–1097 CrossRef CAS PubMed.
- S. Gupta, B. Schade, S. Kumar, C. Böttcher, S. K. Sharma and R. Haag, Small, 2013, 9, 894–904 CrossRef CAS.
- H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325–384 CAS.
- E. Vanbiervliet, S. Fouquay, G. Michaud, F. Simon, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2017, 50, 69–82 CAS.
- Y. Inoue, K. Matsumoto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1076–1081 CAS.
- Y. Chen, B. Chen and J. M. Torkelson, Macromolecules, 2023, 56, 3687–3702 CAS.
- A. K. Vaish, S. Consul, A. Agrawal, S. C. Chaudhary, M. Gutch, N. Jain and M. M. Singh, J. Emerg. Trauma Shock, 2013, 6, 271–275 CrossRef.
- A. Sikder, S. Chakraborty, P. Rajdev, P. Dey and S. Ghosh, Acc. Chem. Res., 2021, 54, 2670–2682 CrossRef CAS.
- S. Kumari, K. Achazi, P. Dey, R. Haag and J. Dernedde, Biomacromolecules, 2019, 20, 1157–1166 CrossRef CAS PubMed.
- J. Khandare, A. Mohr, M. Calderón, P. Welker, K. Licha and R. Haag, Biomaterials, 2010, 31, 4268–4277 CrossRef CAS PubMed.
- M. Wyszogrodzka and R. Haag, Langmuir, 2009, 25, 5703–5712 CAS.
- M. Wyszogrodzka, K. Möws, S. Kamlage, J. Wodzińska, B. Plietker and R. Haag, Eur. J. Org. Chem., 2008, 2008, 53–63 Search PubMed.
- G. Chen, J. Kumar, A. Gregory and M. H. Stenzel, Chem. Commun., 2009, 6291–6293 CAS.
- M. J. Robb, D. Montarnal, N. D. Eisenmenger, S.-Y. Ku, M. L. Chabinyc and C. J. Hawker, Macromolecules, 2013, 46, 6431–6438 CAS.
- P. Dey, T. Bergmann, J. L. Cuellar-Camacho, S. Ehrmann, M. S. Chowdhury, M. Zhang, I. Dahmani, R. Haag and W. Azab, ACS Nano, 2018, 12, 6429–6442 CrossRef CAS.
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS.
- P. Rajdev and S. Ghosh, J. Phys. Chem. B, 2019, 123, 327–342 CrossRef CAS PubMed.
- P. Dey, P. Rajdev, P. Pramanik, R. Haag and S. Ghosh, Macromolecules, 2020, 53, 7044–7052 CrossRef CAS.
- Y. Wang, C. M. Kausch, M. Chun, R. P. Quirk and W. L. Mattice, Macromolecules, 1995, 28, 904–911 CAS.
- S. Holappa, L. Kantonen, F. M. Winnik and H. Tenhu, Macromolecules, 2004, 37, 7008–7018 CAS.
- W. L. F. Armarego, in Purification of Laboratory Chemicals, ed. W. L. F. Armarego and C. L. L. Chai, Butterworth-Heinemann, Oxford, 6th edn, 2009, p. xv Search PubMed.
- I. N. Kurniasih, H. Liang, S. Kumar, A. Mohr, S. K. Sharma, J. P. Rabe and R. Haag, J. Mater. Chem. B, 2013, 1, 3569–3577 CAS.
- R. Kilic Boz, D. Aydin, S. Kocak, B. Golba, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2022, 33, 839–847 CAS.
- F. Calik, A. Degirmenci, R. Sanyal and A. Sanyal, Eur. Polym. J., 2023, 201, 112548 CrossRef CAS.
- A. Degirmenci, G. Yeter Bas, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2022, 33, 1672–1684 CrossRef CAS.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting data and experimental section. See DOI: https://doi.org/10.1039/d4py01028k |
| This journal is © The Royal Society of Chemistry 2025 |