
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoacervation for biomedical applications: innovations involving nucleic acids
Kimiasadat
Mirlohi
 and
Whitney C.
Blocher McTigue
*
and
Whitney C.
Blocher McTigue
*
Department of Chemical and Biomolecular Engineering, Lehigh University, Bethlehem, PA 18015, USA. E-mail: whb322@lehigh.edu
First published on 6th December 2024
Abstract
Gene therapies, drug delivery systems, vaccines, and many other therapeutics, although seeing breakthroughs over the past few decades, still suffer from poor stability, biocompatibility, and targeting. Coacervation, a liquid–liquid phase separation phenomenon, is a pivotal technique increasingly employed to enhance the effectiveness of therapeutics. Through coacervation strategies, many current challenges in therapeutic formulations can be addressed due to the tunable nature of this technique. However, much remains to be explored to enhance these strategies further and scale them from the benchtop to industrial applications. In this review, we highlight the underlying mechanisms of coacervation, elucidating how factors such as pH, ionic strength, temperature, chirality, and charge patterning influence the formation of coacervates and the encapsulation of active ingredients. We then present a perspective on current strategies harnessing these systems, specifically for nucleic acid-based therapeutics. These include peptide-, protein-, and polymer-based approaches, nanocarriers, and hybrid methods, each offering unique advantages and challenges. Nucleic acid-based therapeutics are crucial for designing rapid responses to diseases, particularly in pandemics. While these exciting systems offer many advantages, they also present limitations and challenges which are explored in this work. Exploring coacervation in the biomedical frontier opens new avenues for innovative nucleic acid-based treatments, marking a significant stride towards advanced therapeutic solutions.
1. Introduction
Complex coacervation is a process of liquid–liquid phase separation that occurs in solutions of oppositely charged macromolecules,1 such as proteins,2 polysaccharides,2 polynucleotides,3 synthetic polymers,4 surfactants,5 and their combinations.6–8 The driving force for coacervation is the electrostatic attraction between the charged groups of the macromolecules,2,9,10 which is balanced by the entropic effects of the release of counterions and water molecules from the macromolecular complexes.11–13 The resulting coacervate phase consists of microenvironments with a concentrated mixture of the interacting macromolecules without the need for membranes,14 while the supernatant phase is depleted of them.15,16 Coacervation was first observed by Bungenberg de Jong and Kruyt in the early 1900s in solutions of gelatin and gum arabic,17 and since then it has been extensively studied and applied in various fields, such as food science,18–21 personal care products,22–24 biotechnology,24,25 medicine,26–29 and materials science.26,30,31 Coacervation can be used to encapsulate, protect, and deliver biomolecules,32,33 such as proteins,34–37 enzymes,7,38 and nucleic acids,36,39–41 as well as to create self-assembled structures, such as micelles,38,42–44 vesicles,45,46 and fibers.47,48 The properties and applications of coacervates depend on the nature, composition, and ratio of the macromolecular components,49–51 as well as on the environmental factors, such as salt concentration,42,52,53 pH,54–56 temperature,57–59 and additives.5,60,61In the biomedical field, coacervation is significant for its ability to encapsulate biomolecules, thereby enhancing their kinetic stability and delivery.62 Encapsulation is particularly important for nucleic acid-based therapeutics, which are otherwise chemically unstable and vulnerable to enzymatic degradation63 and have difficulties passively crossing the negatively charged biological membranes.64 The importance of nucleic acid encapsulation extends beyond mere protection; it is a pivotal step in the stabilization of therapeutic molecules, which are often subject to the rigors of the cold supply chain.65 The cold chain ensures precise temperature control to prevent degradation, which can significantly increase the cost and complexity of delivering these sensitive treatments, especially in resource-limited settings.66 Coacervation-based encapsulation offers a promising avenue in this regard. By creating microenvironments that shield nucleic acids from thermal fluctuations and enzymatic activity, as well as allowing the potential for controlled release of the therapeutic cargo,67,68 coacervation can potentially reduce or eliminate the need for cold chain logistics, thereby simplifying the distribution and storage of nucleic acid-based therapeutics. This could lead to more equitable healthcare delivery and a broader reach of life-saving treatments, particularly in areas where cold chain infrastructure is lacking or non-existent.
In this review, we provide a perspective on coacervation as a pivotal technique for nucleic acid delivery. We delve into the factors that influence coacervation processes and examine the various encapsulation methods that harness peptides, proteins, and polymers. The applications of coacervation in gene therapy, drug delivery systems, and vaccines will be highlighted, alongside a critical analysis of the challenges and limitations inherent to these approaches. Finally, we cast a forward-looking gaze at the future perspectives and research directions, discussing advanced coacervation techniques, targeted delivery systems, and the burgeoning potential of combination therapies. This review aims to provide a thorough understanding of the current landscape and the exciting possibilities in the realm of nucleic acid delivery.
2. Coacervation mechanisms and complex morphology
The complex coacervation process is a fascinating phenomenon predominantly occurring in colloidal systems, where two or more oppositely charged polymers interact in solution to form a dense, polymer-rich phase known as a coacervate, alongside a dilute polymer-poor phase referred to as the supernatant.4 This phase separation is driven by electrostatic interactions between the charged groups of the polymers, leading to the aggregation and subsequent phase separation from the bulk solution. The mechanism behind complex coacervation involves a delicate balance of intermolecular forces, including electrostatic interactions, hydrogen bonding, and van der Waals forces, countered by the entropy-driven tendency of the system to remain mixed4,69 (Fig. 1(A)). The entropic gains from the release of counterions are another significant driving force for phase separation.70 In coacervation, while the oppositely charged polymers are aligning into condensates and therefore losing their local entropy, counterions are gaining their translational entropy by getting released into the supernatant.70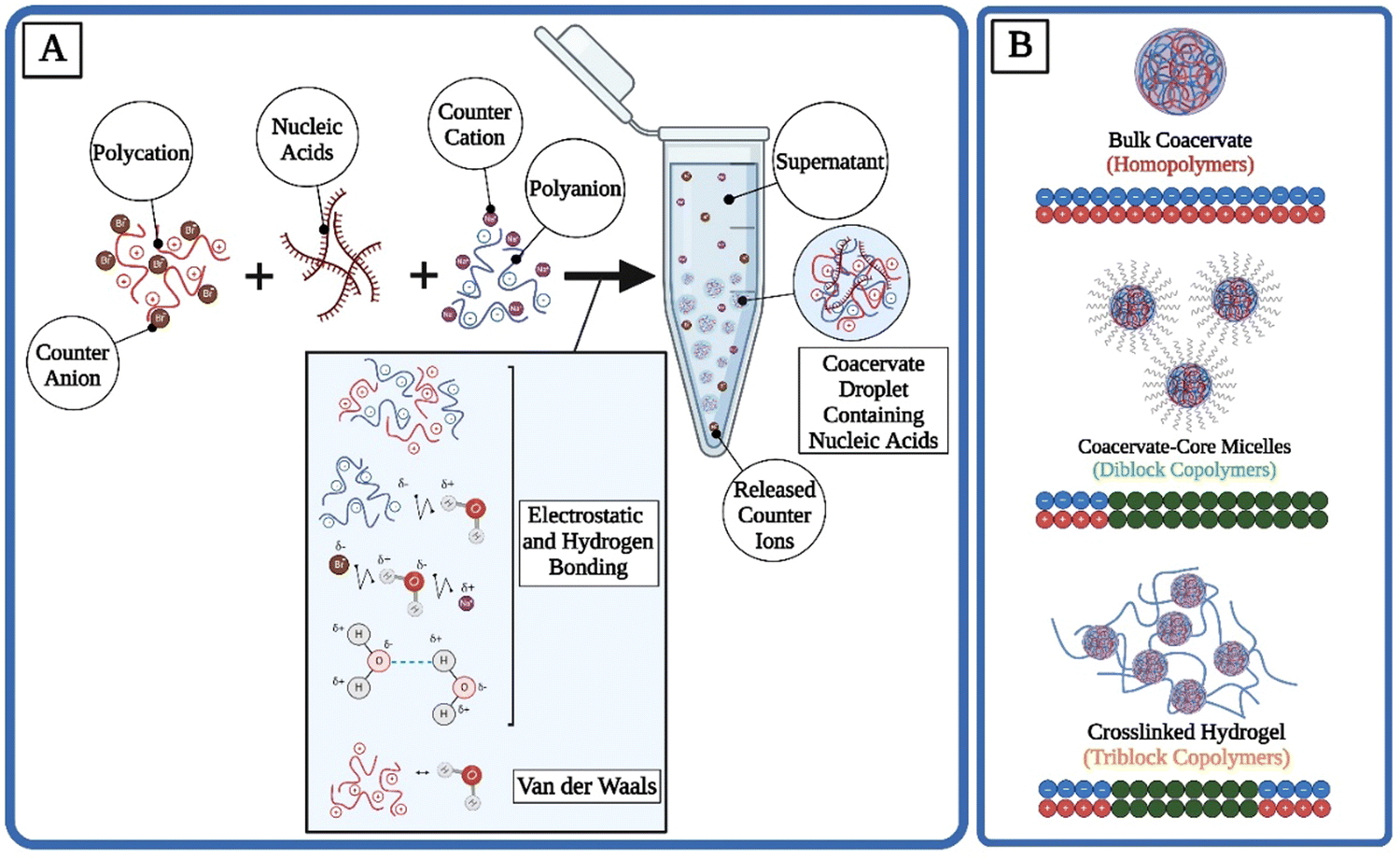 | ||
| Fig. 1 Coacervation mechanisms and the effect of polymer structure on complex morphology. (A) In complex coacervation, polycations paired with negatively charged counter ions (e.g., bromine) can interact with polyanions paired with positively charged counter ions (e.g., sodium). Electrostatic interactions, hydrogen bonding, and van der Waals interactions are possible among all components within the aqueous solution. The dense coacervate droplets have high concentrations of the oppositely charged polyelectrolytes and encapsulate the nucleic acids, while the counter ions are released into the supernatant and help drive phase separation with the entropic gains of their release. (B) Homopolymers made of charged monomers form bulk complex coacervate droplets, di- and tri-block copolymers consisting of charged and uncharged sections can lead to coacervate-core micelles and crosslinked hydrogels, respectively (Created in BioRender. Mirlohi, K. (2024) https://BioRender.com/l47c078). | ||
Depending on the structure and charge patterning of the polyelectrolytes within the system, various morphologies are possible for coacervates. A mixture of homopolymers leads to bulk phase separation.42,67,71 The addition of a hydrophobic block to the polyelectrolyte can cause the formation of micelles with coacervate corona structures.42,72 If hydrophilic but uncharged blocks (such as poly(ethylene glycol) (PEG)) are added to polyelectrolytes, coacervate-core micelles (C3Ms) become possible instead.73–75 Hydrogel-like coacervate networks can be formed under higher concentrations of tri-block copolymers.67,76,77 Although many complex morphologies are possible, here the structure and applications of bulk coacervate droplets, C3Ms, and coacervate-based hydrogels are highlighted (Fig. 1(B) and Table 1).
| Morphology | Polymers | Biomedical application example | Advantages | Disadvantages |
|---|---|---|---|---|
| Bulk coacervate | Homopolymers | Coating material for cell culture and biomimetic adhesives,78 protocells,79 bioimaging80 | Simple preparation,69 easier characterization81 | Limited control over structure, the potential for coalescence, and instability69 |
| Coacervate-core micelles | Di-block copolymers | Drug delivery, bioimaging82 | High stability, therapeutically preferred size (10–100 nm),83–85 promoted cellular uptake,86 facilitated endosomal escape, tunable properties86 | Challenging synthesis, the potential for aggregation86 |
| Coacervate-based hydrogels | Tri-block copolymers | Tissue engineering,87 wound healing88 | High mechanical strength, flexibility, and tunable degradation rates89 | Challenging crosslinking process,87 potential for cytotoxicity89 |
Bulk coacervate droplets are usually not stable structures for drug delivery as they coalesce after some time.90,91 On the other hand, micelles, specifically polymeric micelles, are commonly employed structures as therapeutic carriers due to their stability in low concentrations, their clinically relevant size of 10–100 nm,83 their ability to encapsulate and deliver hydrophobic drugs within their hydrophobic core, the opportunity to functionalize their surface for better targeting, and, lastly, their potential for concurrent delivery of multiple active ingredients92 (Table 1). These versatile micelles can also incorporate coacervates within their core. For example, in a study by Vogelaar and coworkers, an antibiotic called colistin was complexed with poly(ethylene oxide)-b-poly(methacrylic acid) (PEO-b-PMAA) block copolymers to form C3Ms.93 Colistin is used as a last resort treatment against Gram-negative bacterial infections, and their work showed decreased enzymatic degradation after complexation. Based on the entropy-driven mechanisms shown in Fig. 1, polymeric micelles can be prepared solely through the reduction of free energy.92 However, it must not be concluded that enthalpy never plays a role. For example, under diluted conditions, polymeric micelles that have formed solely based on entropy gains can dissociate, making them unfit for parenteral (beyond intestinal) drug delivery applications.92 Although isothermal titration calorimetry (ITC) measurements confirm that coacervation is not dominated by enthalpic effects, they confirm the experimental observations of enthalpy's role in coacervation: enthalpy has a small, positive contribution to coacervation.70
Lastly, coacervate-based hydrogel systems can be used for various drug delivery purposes where other methods might not be as effective, such as for oral ulcers. In their study, Chen and coworkers developed coacervate-based hydrogels composed of Pluronic F68 (F68) and tea polyphenols (TP) and reported that the F68-TP coacervates attached to porcine skin easily.88 They demonstrated that their hydrogel system effectively addresses the issue of poor adhesion seen in conventional drug delivery systems within the moist environment of the mouth, while also accommodating the frequent movements of the lips. In summary, bulk coacervate droplets, micelles, and coacervate-based hydrogels each serve specific purposes in therapeutics. Bulk coacervates, while less structurally stable due to coalescence, are foundational in the study of phase separation. Polymeric micelles stand out for their stability, encapsulation capabilities, and potential for targeted delivery, making them invaluable for drug delivery applications. Lastly, coacervate-based hydrogels are exceptionally suited for localized treatments like oral ulcers, offering improved adhesion and flexibility (Table 1).
3. Factors affecting coacervation
The formation of coacervates can be influenced by several factors essential to understanding the precise design and application of coacervate-based systems. Additionally, the ability to control phase behavior using a multitude of factors makes coacervation a wonderful technology for biomedical applications due to its tunability. Factors such as ionic strength, pH, temperature, and polymer structure and concentration play crucial roles in modulating coacervation.943.1 Environmental effects
For example, amino acids are weak polyelectrolytes known for the shift in their charge profile depending on the pH of their environment.106–108 Amino acids have at least two pKa values, one for the carboxyl group and one for the amino group, but they can have more pKa values depending on their side chains. Therefore, pH controls the overall charge of peptides and polypeptides made of ionizable amino acids.109 Similar principles apply to other polymers consisting of ionizable groups. For many systems, maximum electrostatic interactions and therefore coacervation are observed when the oppositely charged species have balanced charges, corresponding to charge neutralization.9 This means that if two polymers with similar pKa values are studied, they would achieve maximum complexation at balanced charges whereas with off-stoichiometric ratios, they would not experience the same extent of electrostatic affinity (Fig. 2(A)). To achieve both a balance of charges and more possible electrostatic interactions, an option is to use polymers with different pKa values and conduct experiments at a pH away from both of their pKa values, meaning that they would both be fully charged. For example, lysine, with a pKa of approximately 11, and glutamate, with a pKa of approximately 4, exhibit distinct ionization behaviors at physiological pH110 (Fig. 2(B)). The Henderson–Hasselbalch equation elucidates that at pH values ∼2 units away from pKa, the charge of the amino acids will be close to ±1.109,111 At pH 7, lysine's amino group remains protonated, resulting in a positive charge, while glutamate's carboxyl group is deprotonated, giving it a negative charge. Consequently, homopolymers of lysine and glutamate would be fully charged at pH 7. This understanding is essential for predicting the electrostatic interactions and coacervation behavior of polypeptides, as the balanced charges at pH 7 lead to optimal conditions for coacervation.
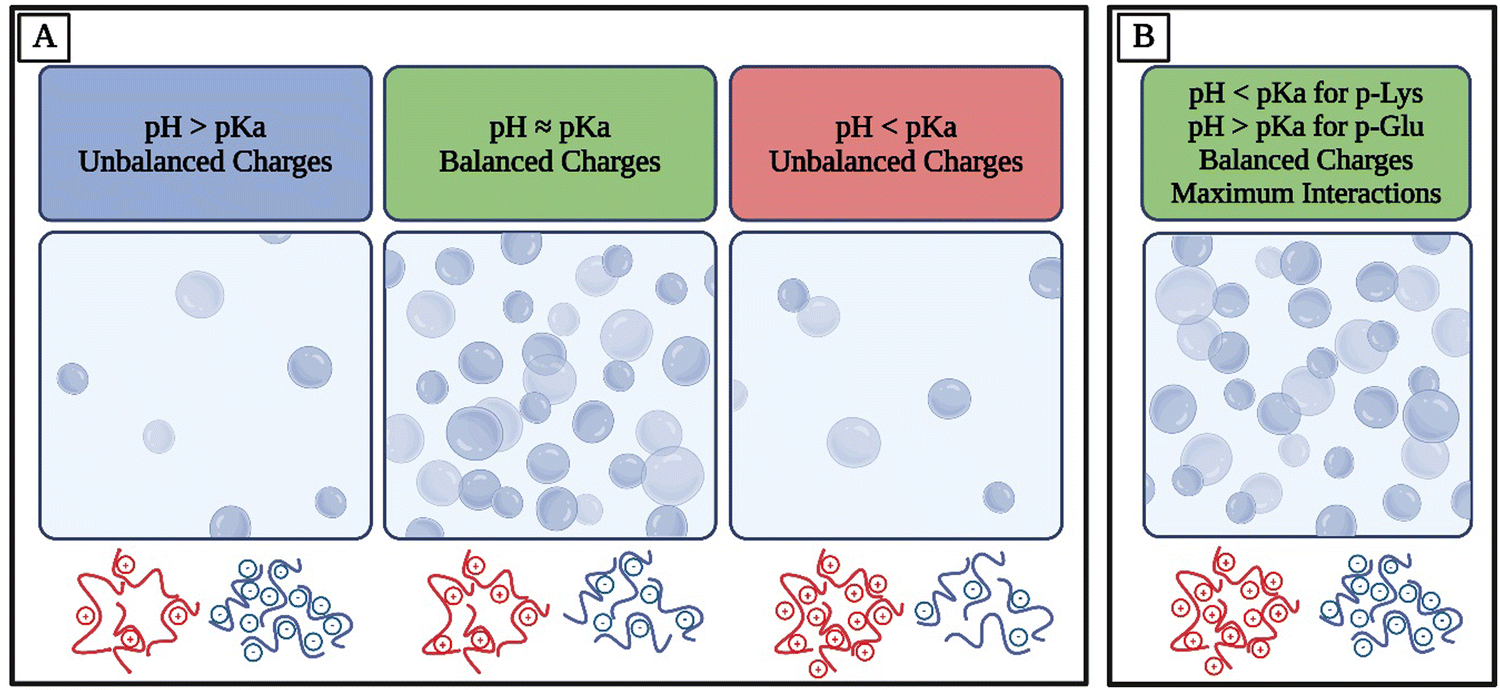 | ||
| Fig. 2 A schematic demonstrating the effect of pH relative to pKa and the balance of charges of the polymers present in the solution on phase separation. The charge density of polymers involved in coacervation depends on the pH relative to their specific pKa values. (A) In the coacervation of polymers with similar pKa values, as the pH approaches the pKa of the polymers, their charges become more balanced, with nearly half of their ionizable charges protonated and the other deprotonated. While the polymers are not fully charged at this pH, their balanced charges improve their electrostatic interactions, consequently leading to phase separation. (B) In the coacervation of polymers with pKa values further apart, the optimal pH for phase separation is away from the pKa of both polymers. At this pH, they are both fully charged, and the opposite charges are also balanced, leading to an increase in electrostatic interactions. Schematics below each box illustrates relative charge of each species (Created in BioRender. Mirlohi, K. (2024) https://BioRender.com/r80p465). | ||
Sensitivity to pH can allow control over the behavior of drug delivery systems that leverage coacervation, where they can release the cargo at specific pH ranges. For example, the targeted treatment of ulcerative colitis has been a challenge due to the wide pH range, and coacervate droplets containing emodin as the active ingredient have shown great promise due to their high stability and pH-responsive release behavior.112 These concepts can be applied to the delivery of nucleic acids as well. In a study, Sun and coworkers designed histine-rich beak peptides (HBpep-K) that were redox- and pH-sensitive.113 They were able to modulate the pH sensitivity of their peptide by conjugating the only lysine residue with a self-immolative moiety (HBpep-SR). At neutral pH, HBpep-K remains dissolved in the solution. However, when the sole lysine residue is modified with a self-immolative group, it can phase-separate into coacervates. In a reducing environment like the glutathione (GSH)-rich cytosol, HBpep-SR undergoes reduction, leading to self-catalytic removal of the SR group. This process causes the coacervate to disassemble. When HBpep-SR forms coacervates near neutral pH, it can encapsulate macromolecular therapeutics such as mRNA.113 Upon exposure to cells, these therapeutic-loaded coacervates can cross the cell membrane through an energy-independent pathway, potentially involving cholesterol-dependent lipid rafts. Once inside the cytosol, the coacervates are reduced by GSH, leading to their disassembly and the release of the therapeutic agents. The pH-responsive behavior of coacervates can also help with modeling membrane-less organelles.114,115
In addition to environmental factors such as pH and temperature affecting phase behavior, the characteristics of coacervate systems can be specifically chosen based on the desired application. The presence of salt ions in the solution affects the ionic strength of the system, and working with the temperature of the solution, they affect phase separation. Certain salts can increase the stability of polymers in the solution by enhancing their solubility which explains the thermo-responsive lower critical solution temperature (temperature below which the components of a mixture are completely miscible in all proportions) of such polymers.119
3.2 Polymer and system effects
The choice and design of monomers and the respective polymers involved in a system affects the possible and potential interactions within that system. For example, the concentration and properties of the present polymers play crucial roles. Polymer concentration determines the viscosity and the extent of the phase separation; higher concentrations favor coacervation by increasing the probability of intermolecular interactions.122 Polymer structure, such as the chirality of monomers and charge patterning of the polymer, can determine the morphology and stability of the complexes formed in each system.123 Charge patterning can dictate the phase behavior of coacervates and shift the binodal phase boundary, providing entropic advantages and influencing the thermodynamics of coacervation.99 Chirality can influence biochemical interactions at a molecular level. In biomedical contexts, chirality can influence protein adsorption, cell adhesion, and subsequent behaviors which are crucial for the design of chiral biomaterials.99 Chirality of polymers also affects the phase behavior of their mixture. Studies show that the presence of racemic polymers provides the opportunity for coacervation, whereas mixing homochiral polymers often yields precipitates and solid aggregates.123–127The length and number of strands of the nucleic acid significantly influence phase separation, hence the stability and formation of coacervates.128 Longer nucleic acids provide more binding sites for electrostatic interactions with the coacervate matrix, enhancing the stability of the encapsulated complex.129 This increased interaction can lead to a more robust coacervate structure, capable of protecting the nucleic acid from degradation. Lebold and Best showed that phase separation is favored more in a 40-nucleotide ssDNA compared to a 20-nucleotide ssDNA, and that this trend appears to hold for DNA lengths several orders of magnitude larger than the 20- and 40-nucleotide strands they used.128 Additionally, Lebold and Best demonstrated that phase separation with dsDNA was favored over the more flexible ssDNA. Their results align with what Wand and coworkers noted in their work, where they found that coacervates composed of β-sheet poly(amino acid)s demonstrated greater protein encapsulation efficiency due to their reduced entropy and flexibility.130 The work of Lebold and Best is investigated in more detail in Section 4.6, where the importance of their findings for hybrid systems that involve nucleic acids and other biomolecules is highlighted.
The sequence of nucleic acids can be pivotal for phase separation as it directly affects the electrostatic interactions based on the charge and polarity of the nucleotides, for example, guanine and cytosine have high charge densities. It is important to note that all the aforementioned factors play a role in the balance of electrostatic interactions, rather than having a clear and established role without any exceptions. To determine an outcome for specific systems, all species involved must be considered. For example, even though the charge density and secondary structure (when present) of nucleic acids seem like inherent factors in changing the encapsulation efficiency of a system, some studies suggest that it is much more complex than that. In their work, Frankel et al. compared an RNA sequence with no known secondary or tertiary structure (HH16 S) with another RNA sequence that has significant secondary structure (HDV E ribozyme).131 They studied these oligonucleotides’ ability to displace ATP in the coacervate phase with PAH and found that there was no apparent dependence on secondary structure. They argued that this could be explained by the strong affiliation of RNA nucleotides with PAH in coacervates due to their multivalency compared to ATP. This is just one example of how convoluted and intertwined factors affecting coacervation can be. To have clear and reliable sets of design rules for coacervate encapsulation systems for nucleic acids, much more research is required, both experimental and computational.
A critical decision in the design of encapsulation systems is the choice between binary and ternary systems. In binary systems, cargo, such as nucleic acids, serves as one of the phase-separating species. Conversely, in ternary systems, nucleic acids are sequestered within the coacervate phase through electrostatic interactions. Both binary and ternary coacervate systems offer distinct advantages. Binary systems are the most prevalent and their formation is more precisely regulated by the stoichiometry of the involved species, ionic strength of the solution (e.g., salt presence), and pH.32,132,133 On the other hand, ternary systems made with polymers of similar length demonstrate higher salt resistance and can form over a wider range of polymer ratios compared to binary systems.132,133 This increase in salt resistance and phase separation being possible at broader species ratios means environmental factors exert less influence on their formation, viscoelastic behavior, and stability.32,132,134 A notable challenge for binary coacervate systems is that not all targeted cargo possesses sufficiently strong charges to bind effectively and remain associated with the oppositely charged species. Ternary systems, however, provide the capability to encapsulate even weakly charged cargo.32,135 These systems are discussed further in Section 5.4.
In summary, the control of coacervation in biomedical applications hinges on a deep understanding of these factors. By manipulating pH, ionic strength, temperature, chirality, and charge patterning, researchers can design coacervate systems with desired properties for specific biomedical purposes. The interplay of these factors determines the efficiency and functionality of coacervates in various biomedical contexts. These parameters collectively dictate the physiochemical environment that governs coacervation. Adjusting these parameters can finely tune the formation, stability, and properties of the coacervates, making them suitable for various applications.136
4. Phase separation strategies involving nucleic acids
Understanding the behavior and properties of complex coacervates, especially those involving nucleic acids, sheds light on the fascinating world of intracellular organization and function, as well as developing therapeutics and drug delivery systems. The efficient delivery of nucleic acids to target cells in vivo presents a formidable challenge.137,138 These molecules, crucial for gene therapy and other therapeutic applications,63,139 face rapid degradation in biological media140,141 and swift clearance from circulation.138,142 To fully harness their therapeutic potential, they require sophisticated delivery systems that protect them, facilitate their transport to specific tissues, and ensure their functional integrity.4.1 Incorporating peptides
Peptide-based coacervation relies on the self-assembly of peptides to form coacervates. This method benefits from the functional diversity of amino acids, allowing for the design of peptides with tailored phase separation properties and selective uptake of nucleic acids. In a study by Vieregg and coworkers, the interactions between DNA and polypeptides were explored.143 They observed the effects of salt concentration and temperature on the phase separation of coacervate systems involving single-stranded DNA (ssDNA), double-stranded DNA (dsDNA), and poly(L-lysine) (pLys). They also experimented with the influence of the number of nucleotides (nt) and base pairs (bp) in their ssDNA and dsDNA respectively, as well as the number of amino acids in their pLys. Their studies showcase the tunability of peptide-nucleic acid coacervates comprehensively (Fig. 3).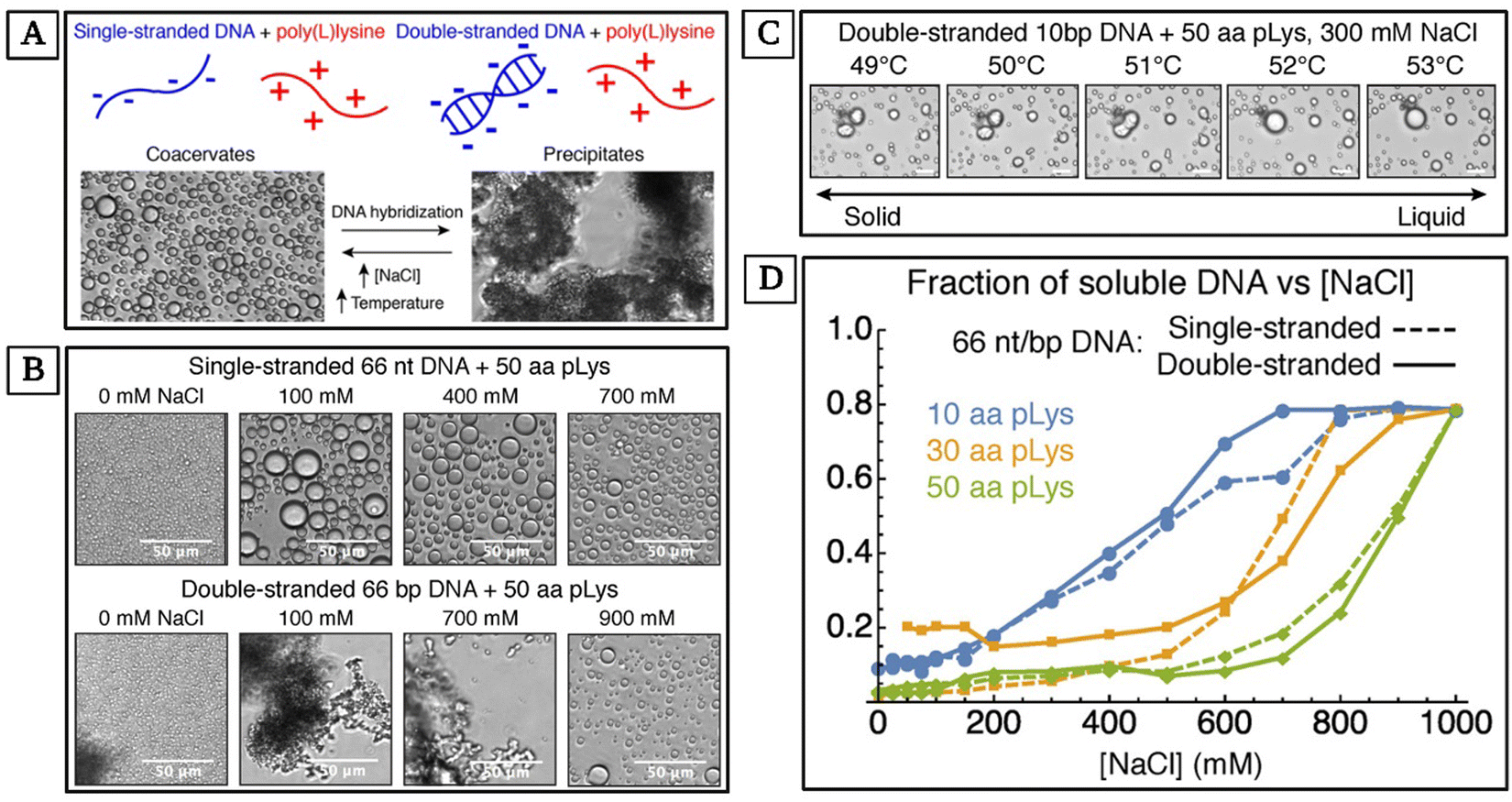 | ||
| Fig. 3 Phase control of oligonucleotide–peptide complexes. (A) A summary of complex behavior based on the structure of DNA, concentration of salt, and temperature. (B) The effect of salt concentration on complexes between poly(L-lysine) (pLys) and 66 nt ssDNA, and pLys and 66 bp dsDNA. Oligonucleotides are released into the solution as the complex size decreases, but the loss during the precipitate-coacervate is small. (C) Complexes between 10 bp dsDNA and pLys at 300 mM NaCl. The mixture undergoes a melting transition at ∼51 °C. Solid complexes with round edges are observed just below this melting transition temperature and spherical liquids are observed at higher temperatures. (D) The change in the solubility of 66 nt/bp DNA when mixed with 10, 30, and 50 aa pLys, as NaCl concentration increases from 0 to 1000 mM. (Reproduced from ref. 143 with permission. Copyright 2018 American Chemical Society.) | ||
The complex coacervation of dsDNA and polycations is less broadly studied due to precipitation, in contrast to the interactions between more flexible nucleic acids such as ssDNA and polycations which lead to liquid droplets (Fig. 3(A)). By exploring this less-studied case, Vieregg and coworkers showed that short dsDNA and pLys can escape precipitation.143 For their observed complex phase diagram with liquid crystal (LC) mesophases in bulk dsDNA, they argue that short dsDNA supramolecular aggregation and packing in the dense coacervate phase are the main phase behavior regulating parameters. Their work provides a deeper understanding of LC-coacervates which in turn may demystify structures and phase transition mechanisms in biomolecular condensates. Additionally, understanding these mechanisms can help with the design of stimuli-responsive synthetic self-assembled compartments, exploiting the prebiotic evolution of nucleic acids and peptides.143 Vieregg et al. used bright-field and polarized transmitted optical microscopy to observe the phase behavior of a charge-balanced sample at different salt concentrations. At [NaCl] ≤ 700 mM, they only observed precipitate-like birefringent structures (Fig. 3(B)). At 800 mM ≤ [NaCl] ≤ 900 mM, fluid coacervate droplets formed (Fig. 3(B)). They showed the dehydration and rehydration of the coacervate LC mesophase transitions and observed the mixtures melting at ∼51 °C (Fig. 3(C)). Additionally, they studied the changes in the solubility of 66 nt ssDNA and 66 bp dsDNA complexed with 10, 30, and 50 amino acid pLys as they increased [NaCl] from 0 to 1000 mM. The transition to a single-phase state at higher salt concentrations was inconsistent for mixtures with ssDNA or dsDNA. However, for all systems, phase separation disappeared at NaCl concentrations of 1 M or higher, resulting in a uniform phase (Fig. 3(D)).
Coacervation has also been instrumental in advancing gene therapy by providing a means to protect and deliver genetic material effectively.144,145 Peptide-based coacervates have been utilized for their high loading efficiency and excellent biocompatibility, which are essential for the delivery of therapeutic genes.146,147 These coacervates facilitate the modulation of gene expression and have been applied in the treatment of various biological diseases.146,148–151 The CRISPR gene therapy paradigm, in particular, has benefited from coacervation techniques, which have improved the delivery and stability of CRISPR/cas9 components, thus enhancing the precision and efficiency of gene editing36 (Fig. 4). For example, Abbasi and coworkers were able to co-encapsulate Cas9 mRNA and guide RNA in coacervates for genome editing in mouse brain, demonstrating the potential of coacervation for in vivo genome editing.152
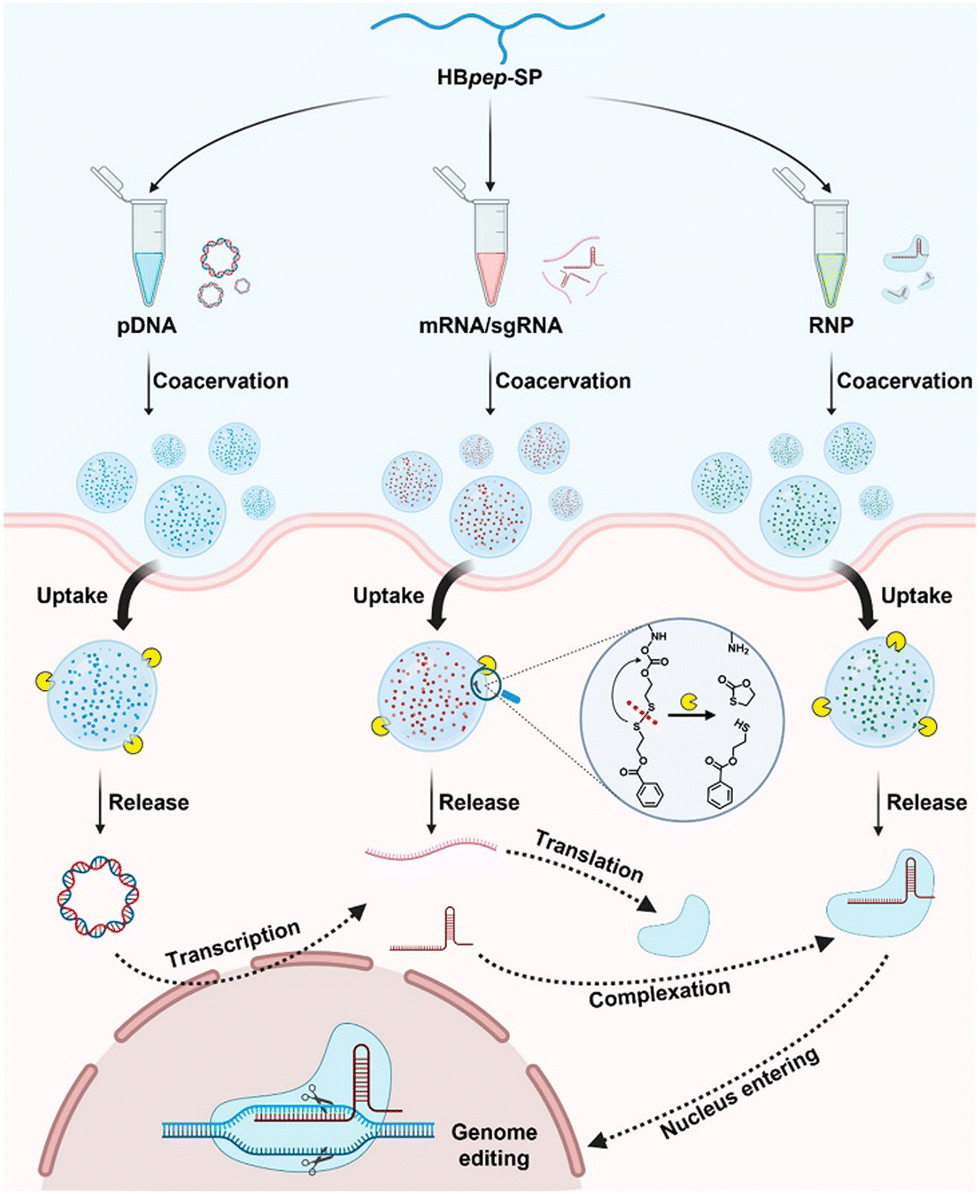 | ||
| Fig. 4 Schematic presentation of universal delivery vehicle for CRISPR/Cas9 genome editing machineries. These machineries are mediated by a redox-responsive peptide (HBpep-SP) that undergoes LLPS due to changes in the pH. The coacervation, uptake, release, and genome editing mechanisms of plasmid DNA (pDNA), messenger RNA (mRNA), single guide RNA (sgRNA), and ribonucleoprotein (RNP) are shown (adapted from ref. 36 with permission. Copyright 2023 American Chemical Society). | ||
A recent study by Aman et al. demonstrated the use of gamma-modified peptide nucleic acids to facilitate targeted DNA invasion, showcasing the potential of peptide-based coacervation in gene editing technologies.153 Aman and coworkers explored the integration of functional peptides into nucleic acid-based nanostructures and highlighted the enhanced biological activity and emerging properties achieved through their approach.153 Their findings confirmed that T7 endonuclease I (T7EI, a structure-selective enzyme) can efficiently cleave DNA invaded by PNAs and demonstrated the potential of their system for targeted DNA cleavage.
4.2 Incorporating proteins
Protein-based coacervation is a nature-inspired approach for compartmentalization and protection of molecules such as nucleic acids. A factor making proteins great molecules for forming complex coacervates with nucleic acids is the diversity of possible interactions between these molecules.154 In a study, Kaushik and coworkers examined the interpolymer interactions between elastin and DNA.155 Interestingly, they found that complex coacervation was possible even when the concentrations of both elastin and DNA were extremely low (between 1 and 35 ppm). They argue that this phenomenon is explained by the ubiquity of DNA–protein interactions that can lead to self-assembled phases. This makes these systems extremely useful in biological applications, as high concentrations of biomolecules such as nucleic acids may not always be easily available.In a study by Zhang and coworkers, the ability to store tau protein reversibly in complex coacervates with RNA was explored.156 Tau is a neuronal protein that forms aggregates in the form of insoluble fibers through self-assembly in several neurodegenerative diseases such as Alzheimer's disease. Additionally, it was observed that in human neuronal cell culture, tau selectively binds to tRNA.156 Zhang et al. discovered a state where many tau molecules maintain their solubility and native-like conformation in protein-rich coacervate droplets with RNA as the negatively charged constituent, which were shown to coalesce in vitro.156 Their work demonstrated that tau-RNA droplets are responsive to changes in temperature and salt concentration. Consequently, these results showed the physiochemical properties of tau that may cause it to have changes associated with neurodegenerative disease.
4.3 Incorporating polymers
Polymer-based coacervation involves the use of polymers to encapsulate nucleic acids, offering protection against enzymatic degradation and facilitating cellular uptake. The design of polymer-based nanoparticles has been a focus of recent research, aiming to ensure targeted delivery and controlled release of therapeutic nucleic acids.100,157,158 Innovations in this field have led to the development of novel delivery systems for a broad range of infectious, chronic, and genetic disorders.159–162Frankel et al. studied the complex coacervation of poly(allylamine hydrochloride) (PAH, 15 kDa) with anionic nucleotides adenosine 5′-mono-, di-, and triphosphate (AMP, ADP, ATP).131 In their work, they explored the effect of PAH and nucleotide concentrations on the turbidity of solutions. They also measured turbidity as a function of PAH concentration with 2.5 mM ATP, 3.3 mM ADP, and 5 mM AMP (Fig. 5(I)). Additionally, they acquired optical micrographs of PAH/nucleotide coacervate droplets, where they mixed 38.5 μM PAH, 2 mM HEPES at pH 7, and with 5 mM Mg2+ (Fig. 5(II)). From their turbidity studies, they concluded that the coacervation between the nucleotides and PAH was maximal near charge neutralization which is consistent with previous studies.
 | ||
| Fig. 5 Comparison of turbidity of solutions as a function of nucleotide and PAH concentration across different nucleotides, along with optical micrographs of these systems to highlight complex morphology. (I, A) Turbidity with 38.5 μM PAH as a function of nucleotide concentration. (I, B) Turbidity as a function of PAH concentration with 2.5 mM ATP (black squares), 3.3 mM ADP (red triangles), and 5 mM AMP (blue circles), all in 5 mM Mg2+ and 2 mM HEPES at pH 7. (II) Optical micrographs of PAH/nucleotide complexation prepared with 38.5 μM PAH, 2 mM HEPES pH 7, and 5 mM Mg2+. (II, A–C) Solutions made with equal charge concentration of 10 mM negative and positive moieties with (A) ATP, (B) ADP, and (C) AMP. (II, D–F) The concentration of (D) ATP, (E) ADP, (F) and AMP were kept constant at 10 mM. Micrographs in II, A–E, show coacervation, while II, F shows an aggregate system. Scale bars represent 15 μm (Reproduced from ref. 131 with permission. Copyright 2016 American Chemical Society). | ||
The greater capacity of ATP and ADP to counteract PAH compared to AMP is evident in the broader range of PAH concentrations that exhibit liquid–liquid phase separation for the more strongly charged nucleotides.131 When nucleotides were present at higher concentrations, samples sustained their turbidity at higher polyamine concentrations up to at least 100 μM PAH. Their work also suggested nucleotide self-association (Fig. 5(I)). They observed AMP-containing aggregates at 10 mM, which they argue might be due to poor packing of the solid particulates, which even after centrifugation did not show any compression (Fig. 5(II)).
A notable system that incorporates polymers in a coacervate delivery system is the use of chitosan, a natural biocompatible polysaccharide, to form DNA nanoparticles for oral allergen-gene immunization. In a study focused on peanut antigen-induced murine anaphylactic responses, Roy and coworkers showed that oral administration of chitosan–DNA nanoparticles led to gene expression in the intestinal epithelium.163 Mice treated with nanoparticles containing a dominant peanut allergen gene (pCMVArah2) produced secretory IgA and serum IgG2a. Compared to non-immunized mice or those treated with naked DNA, the immunized mice exhibited a significant reduction in allergen-induced anaphylaxis, as evidenced by lower levels of IgE, plasma histamine, and vascular leakage. Their findings highlight the potential of polymer–DNA nanoparticles in modulating immune responses, especially notable for the biocompatibility of natural polymers such as chitosan.
In another study, Nishida and coworkers synthesized ligand-installed block copolymers for systematic delivery of siRNA, successfully silencing the E6 and E7 human papillomavirus (HPV) oncogenes.164 They created micelles with ligand-installed, functionalized cRGD-PEG-PLK and siRNA, specifically designed to target the cancer cell surface. Their results from mice treated with these micelles for tumors demonstrated the great therapeutic potential of these systems against HPV-associated cancer. Other groups have incorporated uncharged functional polymers in poly-lysine/pDNA to make ternary complexes and achieved enhanced in vitro gene transfection without cytotoxicity, and take a step toward developing innovative gene and drug delivery systems.165 In addition to delivery advantages provided by polymeric encapsulation systems for nucleic acids, some studies have focused on the potential of polymeric complexes in protecting nucleic acids and therapeutic proteins against freezing, heat, and freeze-drying stress.166
4.4 Nucleic acids in hybrid strategies and beyond
A great characteristic of coacervate encapsulation and delivery systems for nucleic acids is the ability to tune their properties by fine control over their structure and contents.132,167 Therefore, it is possible to leverage the useful properties of various biomolecule groups at once, in the form of hybrid systems.The surface charge of delivery vehicles plays an important role in cellular uptake and transfection efficiency.168–170 To overcome this challenge, Li and coworkers proposed hybrid PEGylated nanoparticles (HNPs) in their study.171 To provide colloidal stability, they included an outer layer of poly(ethylene glycol) in the form of poly(ethylene glycol)-b-poly(aspartate)-adamantane (PEG-P(asp)-Ad). Their HNPs also included poly(ethyleminine)10k (PEI10k) which forms complex coacervate with poly(asp) and prevents premature dissociation. Lastly, they decorated the PEI10k portion with cyclodextrin (PEI10k-CD) to form the core with reporter plasmid DNA (pDNA). Their HNPs demonstrated enhanced in vitro transfection and increased stability compared to traditional PEGylated nanoparticles (PEG-NP). Their study of intratumoral injections compared the delivery efficiencies of HNPs, PEG-NP, and PEI25k, and highlighted the success of HNPs in delivering pDNA into tumors. They argue that this enhanced efficiency is a result of the PEG shell and coacervation integration at the interface of PEI10k-CD/pDNA core compared to PEG-NP and PEI25k.
In another study, Cai et al. studied coacervate systems involving gold nanoparticles (AuNPs) functionalized with elastin-like peptides (ELPs) as potential delivery systems.172 Their AuNPs were functionalized with a cysteine-terminated 96-repeat of the VPGCG sequence (V96-Cys). ELPs are known to be thermo-responsive,173 which allowed for controlling the size of these particles from 250 to 930 nm.172 Their findings were that ELP-decorated AuNPs form clusters in a coacervate phase in the presence of free unimers (single chains of V96-Cys) above the transition temperature (Tt). These clusters in the coacervate phase can then release their entrapped cargos below Tt.172 They demonstrated the possibility of precise control over the amounts of hydrophobic molecular cargos, specifically the dye Nile red (NR) and tetracycline (TC) which is a broad-spectrum antibiotic, encapsulated during coacervation, and subsequently released below Tt during deconstruction. As predicted, Cai and coworkers observed the inhibition of cell growth with increased cluster size.
The hydrophobicity of TC is specifically important as their enhanced bioavailability is explored.172 Their results show an avenue for rapidly releasing precise amounts of cargo from size-controlled clusters in the coacervate phase, in addition to being responsive to environmental factors. This can have remarkable applications in drug delivery, combination photothermal therapy, and theranostic applications.172 The creative approach to nucleic acids as functionalizing groups rather than cargo itself brings about even more opportunities to leverage various properties of these biomolecules.
The findings of Lebold and Best regarding the formation of coacervates in mixtures of nucleic acids and histones agree with experimental results. Leicher and coworkers explored the binding and coacervation of nucleic acids by linker histone H1.174 Their work provides a more nuanced perspective on what role H1 plays in genome organization and maintenance while providing insight into the effects of single- or double-strandedness of nucleic acids on how they interact with H1 and their assembly dynamics. They found that ssDNA has a higher absorbance index compared to dsDNA, indicating more complexation (Fig. 6(A) and (B)). Additionally, they investigated the effect of the size of the nucleic acid chain on absorbance for the mixture of H1 with ssDNA, dsDNA, and RNA (Fig. 6(C)–(E)). They found that longer chains of ssDNA yield higher turbidity readings when complexing with H1 (Fig. 6(C)) and confirmed that ssDNA has higher turbidity values compared to dsDNA with the same sequence and length (Fig. 6(D)). Additionally, they observed the same pattern of increasing turbidity values with increasing length when studying RNA mixtures with H1 instead of ssDNA and dsDNA (Fig. 6(E)).
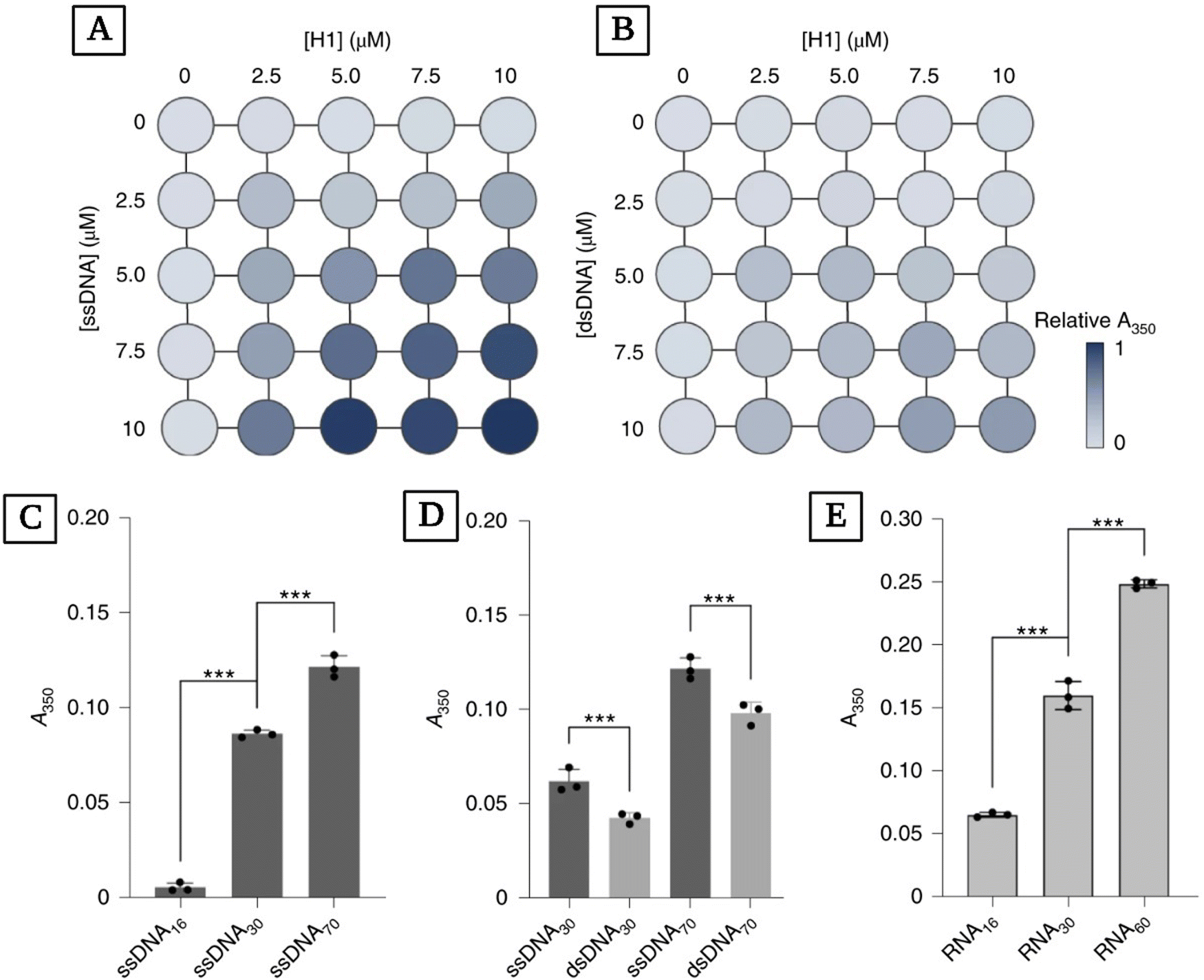 | ||
| Fig. 6 Studying the complexation of H1 with ssDNA, dsDNA, RNA, and the effects of nucleic acid length on the turbidity of these mixtures. Matrix representation of turbidity (A350) measured at different concentrations of H1 and ssDNA (A) and dsDNA (B). (C) Turbidity readings for mixtures of full-length H1 with ssDNA of different lengths. ssDNA concentrations were normalized to yield the same total number of nucleotides (44 μM ssDNA16, 23 μM ssDNA30, 10 μM ssDNA70). (D) Turbidity values for H1 mixed with ssDNA and dsDNA of the same length and sequence. The concentrations of nucleic acids were normalized to yield the same number of nucleotides (10 μM ssDNA30/5 μM dsDNA30 and 10 μM ssDNA70/5 μM dsDNA70). (E) Turbidity values for H1 mixed with different lengths of RNA. RNA concentrations were normalized to yield the same number of nucleotides (44 μM RNA16, 23 μM RNA30, 12 μM RNA60). ***P < 0.001 using one-way analysis of variance (ANOVA) with Tukey's test for multiple comparisons (reproduced from ref. 174 with permission. Copyright 2022 Nature). | ||
 | ||
Fig. 7 Exploring RNA-based coacervates as models for membrane-less organelles. (A) Transmitted light differential interference contrast (DIC) micrographs of bulk polyU/spermine (0.05% polyU and 0.05% spermine weight percent, a charge ratio of 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and polyU/spermidine (0.05% polyU and 0.05% spermidine weight percent, a charge ratio of 61:1). (B, left) Turbidity values versus charge ratio for polyU complexing with spermine (blue) and spermidine (red). (B, right) Turbidity values for 0.05% polyU complexing with 0.5% spermine (blue) or 0.5% spermidine (red) versus 25 mM NaCl concentration at 37 °C. (C) Images of polyU/spermine coacervate dissociation after incubation at 37 °C for 30 min and gradual decrease in temperature starting at t = 0 at a rate of 10 °C min−1 to 10 °C. Droplets are almost completely dissolved after 5 min, and they are fully dissociated after 10 min. Concentrations were 0.05% polyU and 0.5% spermine (a charge ratio of 56:1) or 0.5% spermidine (a charge ratio of 61:1). (D) The partial and entire droplet bleaching studies demonstrate rapid recovery, showcasing the retaining of solute mobility within the coacervate droplets. (E, left) Schematic showing the assembly of lipid vesicles (red circles, not drawn to scale to show individual vesicles) around polyU/spermine coacervates (blue circles). (E, right) DIC and confocal fluorescence images of the polyU/spermine droplets containing fluorescent poly U15, as well as FRAP recovery curve of entire droplet bleaching of the Alexa Fluor 647 poly U15. All scale bars represent 10 μm (reproduced from ref. 178 with permission. Copyright 2016 American Chemical Society). :1) and polyU/spermidine (0.05% polyU and 0.05% spermidine weight percent, a charge ratio of 61:1). (B, left) Turbidity values versus charge ratio for polyU complexing with spermine (blue) and spermidine (red). (B, right) Turbidity values for 0.05% polyU complexing with 0.5% spermine (blue) or 0.5% spermidine (red) versus 25 mM NaCl concentration at 37 °C. (C) Images of polyU/spermine coacervate dissociation after incubation at 37 °C for 30 min and gradual decrease in temperature starting at t = 0 at a rate of 10 °C min−1 to 10 °C. Droplets are almost completely dissolved after 5 min, and they are fully dissociated after 10 min. Concentrations were 0.05% polyU and 0.5% spermine (a charge ratio of 56:1) or 0.5% spermidine (a charge ratio of 61:1). (D) The partial and entire droplet bleaching studies demonstrate rapid recovery, showcasing the retaining of solute mobility within the coacervate droplets. (E, left) Schematic showing the assembly of lipid vesicles (red circles, not drawn to scale to show individual vesicles) around polyU/spermine coacervates (blue circles). (E, right) DIC and confocal fluorescence images of the polyU/spermine droplets containing fluorescent poly U15, as well as FRAP recovery curve of entire droplet bleaching of the Alexa Fluor 647 poly U15. All scale bars represent 10 μm (reproduced from ref. 178 with permission. Copyright 2016 American Chemical Society). | ||
A step forward beyond modeling membrane-less organelles in biological systems is using these structures to manipulate life processes.177 In an effort toward this goal, Zhang and coworkers designed photoactivatable DNA membrane-less organelles using coacervation that were self-stabilizing.177 Specifically, they generated long single-stranded DNA via rolling circle amplification (RCA, an enzymatic process used to amplify circular DNA or RNA molecules179) and used it as the scaffold assembling into membrane-less coacervates. Their DNA membrane-less organelles recruited RCA byproducts and other components which provided for their self-stabilization. Additionally, their condensation had the desired nanoscale size of 10–100 nm.83,177 Their photoactivatable DNA membrane-less organelles accumulated and managed cancer in a mouse model in a spatiotemporal manner. Their results are promising for designing highly stable DNA coacervate membrane-less organelles with controllable bioactivity. This advancement paves the way for developing even more intricate coacervate nucleic acid systems capable of executing sophisticated tasks and regulating complex life processes.
5. Challenges and future explorations
5.1 Stability and delivery
The burgeoning field of coacervation-based delivery systems for nucleic acids is not without its challenges and limitations. Stability issues are at the forefront, as the integrity of coacervates is paramount for the effective delivery of nucleic acids. The lack of a physical membrane in bulk coacervates can lead to rapid coalescence or collapse of the coacervate phases, compromising their structural integrity.180,181 Some methods have been explored to mitigate this challenge. For example, incorporating comb polyelectrolytes in complex coacervates has shown promise in stabilizing phase separation.181 Other groups have explored the effects of mixing flow conditions and polymer composition on coalescence, sedimentation, and aggregation in coacervate systems.182 However, more systematic studies are necessary to formulate the specific factors and their boundaries within coacervate systems for controlling and maintaining phase separation. Another avenue to explore for mitigating the structural maintenance issues is to use di- and tri-block copolymers that are capable of forming more complex and stable structures and systems such as micelles and hydrogels.86Maintaining stability in the complex biological milieu also remains challenging due to factors like pH, salt concentration, and temperature variations.183,184 Additionally, the encapsulated nucleic acids must be protected from enzymatic degradation by nucleases, which is a significant hurdle. While coacervation can enhance the stability of encapsulated cargos, their stability in blood and other biological fluids during storage and upon administration remains a concern.83 Current therapeutics involving nucleic acids would immensely benefit from enhanced kinetic stability, which would increase their shelf life.136 Controlled and sustained release of nucleic acids is also crucial for therapeutic efficacy. The dynamic nature of the biological environment, including interactions with proteins and other biomolecules, can significantly affect release rates. Achieving precise control over release kinetics is complicated by the need to balance stability and permeability.185 Some groups have explored the effects of coacervating species or additives on better control over the release kinetics. For example, Ardestani and coworkers characterized caseinate–pectin coacervates as delivery systems for saffron extract.186 They argued that core/shell and protein/polysaccharide ratios play crucial roles in encapsulation efficiency and loading capacity. Additionally, they argued that the presence of the “guest molecule”, saffron extract, resulted in higher rheological moduli, therefore enhancing thermal, structural, and microstructural properties of the coacervates.186 An area of research in need of further exploration is the effects of additives or “guest molecules” on the properties of coacervates incorporating nucleic acids. Recent studies have utilized molecular dynamics and machine learning to better understand and optimize these mechanisms, however, the variability in biological conditions still poses a significant challenge.7,130
5.2 Immunogenicity and clinical life cycle
Ensuring biocompatibility is essential to avoid adverse immune responses. While coacervates can be formulated from biocompatible materials and even decrease the cytotoxicity of what they’re encapsulating,187 their interaction with cells and tissues must be carefully evaluated to prevent cytotoxicity and immunogenicity.188 The potential for unintended interactions with cellular components can lead to adverse effects, necessitating thorough preclinical testing.146 For example, although poly-lysine has been commonly studied in coacervates for years and has shown great promise,34,189,190 it can cause cytotoxicity at high concentrations.191 Therefore, all coacervate systems must be carefully tested from different perspectives to ensure their safety. Additionally, the scalability and reproducibility of coacervate formulations for clinical applications remain areas of active research. Some significant challenges include manufacturing, as the complexity of coacervate systems can make large-scale production difficult. Ensuring consistent quality and performance across batches is critical for clinical translation. Efficient delivery to target cells and tissues requires overcoming biological barriers such as cellular uptake and endosomal escape. Strategies like surface modification with targeting ligands are being explored to enhance specificity and efficacy,192 but much more enhancement is yet to be achieved.5.3 Further avenues
The landscape of coacervation in nucleic acid delivery is poised for significant advancements, with research efforts converging on the development of advanced coacervation techniques. These novel approaches aim to refine the encapsulation process, enhance the stability of coacervates, and improve the precision of delivery.5.4 Looking onward
The regulatory landscape for nucleic acid-based therapeutics is still evolving. Ensuring compliance with regulatory standards for safety, efficacy, and quality is essential for successful clinical translation. Moreover, there is a need for more explicit experimental exploration of nucleic acids within coacervates. Studies have shown that coacervates can effectively encapsulate and protect nucleic acids, but detailed experimental investigations are required to fully understand the dynamics and interactions within these systems.91 The species involved in coacervation, and their charges greatly influence these systems, as discussed earlier. Whether binary or ternary systems are more effective for nucleic acid delivery, and the charge composition of these molecules for optimal encapsulation and protection against environmental factors and nucleases, require further investigation. For instance, studies have demonstrated enhanced protection of nucleic acids against nucleases when encapsulated within a thermally stabilized coacervate core nanocarrier.39 Moreover, ternary coacervate systems of synthetic polymers have shown greater stability against environmental changes compared to binary systems.132 Combining these findings in a comprehensive study of nucleic acid stability against environmental changes and nucleases could allow us to compare the performance of binary and ternary coacervate systems for nucleic acid delivery.Additionally, understanding the optimal charge composition in ternary systems is essential. Ternary systems with oppositely charged molecules can help encapsulate weakly charged cargos,32,135 raising questions about the behavior of strongly charged molecules within ternary combinations, such as nucleic acids. A ternary system would enhance crowding effects by increasing the number of species involved and provide greater versatility in system properties.15,16,205 However, if another negatively charged molecule is present alongside nucleic acids, it could compete for interactions with positively charged species, potentially increasing the number of free nucleic acid molecules within the system with reduced stability. On the other hand, incorporating two different positively charged molecules could increase electrostatic interaction involving nucleic acids. This approach has been previously applied to nucleic acid aggregates206 and systems without nucleic acids,132 and a comprehensive study applying these insights to nucleic acid encapsulation systems would be highly beneficial. A further necessary consideration is for the interactions of nucleases with each present species. Coacervate systems have been explored as a means to enhance the catalytic activities of enzymes,207,208 which raises questions about design rules that prevent this enhanced enzyme activity when encapsulating nucleic acids to protect them against nucleases.
Recent research has highlighted the potential of peptide-based coacervates as biomimetic protocells, which can sequester and concentrate a wide range of solutes, including nucleic acids.102 These coacervates offer a versatile platform for studying the phase separation and selective uptake of guest molecules, which is crucial for optimizing their use in therapeutic applications.102 Future investigations employing cellular assays can provide insights into cellular uptake, intracellular trafficking, and the overall therapeutic efficacy of the delivery systems.83,209 High-throughput screening platforms, such as RNA-encoded peptide barcodes, have been developed to evaluate the functional delivery of lipid nanoparticles in vivo, which can accelerate the discovery of new technologies for mRNA delivery.209 Additionally, polymeric micelle-based delivery systems have shown promise in enhancing the stability and cellular uptake of nucleic acids, addressing key challenges in their clinical translation.83 These advancements underscore the importance of continued research and innovation in the field to achieve safe and effective nucleic acid-based therapeutics.
6. Conclusion
Coacervation is an innovative mechanism employed in numerous nucleic acid delivery systems for a variety of biomedical applications, including drug delivery and gene therapy. Various factors such as pH, ionic strength, temperature, polymer concentration and structure, the length and sequence of the encapsulated nucleic acids, and the number of species involved in complexation significantly influence and control coacervation-based nucleic acid encapsulation systems. These numerous controlling factors render these encapsulation systems highly tunable, allowing for precise manipulation of the properties of the constituent species and the environmental conditions. However, the necessity to consider such a multitude of factors, coupled with the complexities of the biological milieu and potential immune responses elicited by these delivery vehicles, presents significant challenges for their practical application and large-scale production.Stability issues of the encapsulated nucleic acids and the coacervate phase itself remain a critical concern. The lack of a physical membrane can lead to coalescence or collapse of the coacervate phase, compromising the structural integrity of delivery systems. Additionally, our understanding of the loading and release kinetics of nucleic acids within coacervate systems is still incomplete, complicating the optimization of these delivery vehicles for therapeutic use.
Moreover, there remains a substantial gap in our understanding of coacervate-based encapsulation systems for nucleic acid delivery, particularly regarding their interactions with cells and the body as a whole. This gap encompasses several critical aspects, including the mechanisms by which coacervates interact with cellular membranes, the intracellular trafficking pathways they utilize, and their potential immunogenicity. Understanding these interactions is crucial for optimizing the design of coacervate-based delivery systems to ensure efficient and targeted delivery of nucleic acids while minimizing adverse effects.
Future research should continue to explore hybrid systems that incorporate various materials such as peptides, polymers, proteins, and lipids to harness their combined advantages. For instance, incorporating peptides can enhance cell penetration and targeting, while polymers can provide structural stability and controlled release properties. Proteins can offer specific binding capabilities, and lipids can improve membrane fusion. Such hybrid systems could significantly advance our understanding of the dynamics within these nucleic acid encapsulation systems, leading to more effective and versatile delivery platforms. Additionally, comprehensive cellular assays including studies on cellular uptake, intracellular trafficking, release kinetics, and the biological activity of the delivered nucleic acids are essential to elucidate the cellular interactions and efficacy of nucleic acid loading and delivery, ultimately paving the way for therapeutic applications and clinical translation.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- A. N. Singh and A. Yethiraj, J. Phys. Chem. B, 2021, 125, 3023–3031 CrossRef CAS PubMed.
- C. G. De Kruif, F. Weinbreck and R. De Vries, Curr. Opin. Colloid Interface Sci., 2004, 9, 340–349 CrossRef CAS.
- J. R. Vieregg and T.-Y. D. Tang, Curr. Opin. Colloid Interface Sci., 2016, 26, 50–57 CrossRef CAS.
- C. E. Sing and S. L. Perry, Soft Matter, 2020, 16, 2885–2914 RSC.
- W. Zhao and Y. Wang, Adv. Colloid Interface Sci., 2017, 239, 199–212 CrossRef CAS PubMed.
- T. P. Fraccia and N. Martin, Nat. Commun., 2023, 14, 2606 CrossRef CAS PubMed.
- J. Zheng, P. Van der Meeren and W. Sun, Aggregate, 2024, 5, e449 CrossRef CAS.
- N. Khan and B. Brettmann, Polymers, 2019, 11, 51 CrossRef PubMed.
- E. Kizilay, A. B. Kayitmazer and P. L. Dubin, Adv. Colloid Interface Sci., 2011, 167, 24–37 CrossRef CAS PubMed.
- S. Chen and Z.-G. Wang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209975119 CrossRef CAS PubMed.
- Z. Ou and M. Muthukumar, J. Chem. Phys., 2006, 124, 154902 CrossRef PubMed.
- X. Xu, Q. Ran, P. Dey, R. Nikam, R. Haag, M. Ballauff and J. Dzubiella, Biomacromolecules, 2018, 19, 409–416 CrossRef CAS PubMed.
- M. Ghasemi, S. Friedowitz and R. G. Larson, Soft Matter, 2020, 16, 10640–10656 RSC.
- A. Sathyavageeswaran, J. Bonesso Sabadini and S. L. Perry, Acc. Chem. Res., 2024, 57, 386–398 CrossRef CAS PubMed.
- S. Biswas, A. L. Hecht, S. A. Noble, Q. Huang, R. E. Gillilan and A. Y. Xu, Biomacromolecules, 2023, 24, 4771–4782 CrossRef CAS PubMed.
- A. M. Marianelli, B. M. Miller and C. D. Keating, Soft Matter, 2018, 14, 368–378 RSC.
- P. Koets, J. Phys. Chem., 1936, 40, 1191–1200 CrossRef CAS.
- T. Moschakis and C. G. Biliaderis, Curr. Opin. Colloid Interface Sci., 2017, 28, 96–109 CrossRef CAS.
- L. A. Bosnea, T. Moschakis and C. G. Biliaderis, Food Bioprocess Technol., 2014, 7, 2767–2781 CrossRef CAS.
- B. Wu, B. Degner and D. J. McClements, J. Phys.: Condens. Matter, 2014, 26, 464104 CrossRef PubMed.
- S. Rojas-Moreno, F. Cárdenas-Bailón, G. Osorio-Revilla, T. Gallardo-Velázquez and J. Proal-Nájera, Food Measure, 2018, 12, 650–660 CrossRef.
- A. Ammala, Int. J. Cosmet. Sci., 2013, 35, 113–124 CrossRef CAS.
- C. Lepilleur, J. Mullay and C. Kyler, J. Cosmet. Sci., 2011, 62, 161–177 Search PubMed.
- J. D. Tang, S. R. Caliari and K. J. Lampe, Biomacromolecules, 2018, 19, 3925–3935 CrossRef CAS PubMed.
- D. Magnin, Carbohydr. Polym., 2004, 55, 437–453 CrossRef CAS.
- W. C. Blocher and S. L. Perry, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1442 Search PubMed.
- Ö. Karabıyık Acar, S. Bedir, A. B. Kayitmazer and G. T. Kose, Int. J. Biol. Macromol., 2021, 188, 300–312 CrossRef PubMed.
- K. O. Margossian, M. U. Brown, T. Emrick and M. Muthukumar, Nat. Commun., 2022, 13, 2250 CrossRef CAS PubMed.
- J. George Joy, H. K. Son, S.-J. Lee and J.-C. Kim, J. Ind. Eng. Chem., 2024, 134, 432–447 CrossRef CAS.
- Y. Hong, S. Yoo, J. Han, J. Kim, Y. Lee, Y. Jho, Y. S. Kim and D. S. Hwang, Commun. Chem., 2024, 7, 1–10 CrossRef PubMed.
- M. Li, R. Mirshafian, J. Wang, H. Mohanram, K. A. Ahn, S. Hosseinzadeh, K. V. Pervushin, J. H. Waite and J. Yu, Biomacromolecules, 2023, 24, 4190–4198 CrossRef CAS PubMed.
- E. Ban and A. Kim, Int. J. Pharm., 2022, 624, 122058 CrossRef CAS.
- T. Lu, X. Hu, M. H. I. van Haren, E. Spruijt and W. T. S. Huck, Small, 2023, 19, 2303138 CrossRef CAS PubMed.
- K. A. Black, D. Priftis, S. L. Perry, J. Yip, W. Y. Byun and M. Tirrell, ACS Macro Lett., 2014, 3, 1088–1091 CrossRef CAS PubMed.
- L. Ma, C.-R. Su, S.-Y. Li, S. He, A. Nag and Y. Yuan, Food Hydrocolloids, 2023, 144, 108907 CrossRef CAS.
- Y. Sun, X. Xu, L. Chen, W. L. Chew, Y. Ping and A. Miserez, ACS Nano, 2023, 17, 16597–16606 CrossRef CAS PubMed.
- E. Hasanvand and S. M. A. Razavi, Int. Dairy J., 2023, 145, 105716 CrossRef CAS.
- R. Kembaren, R. Fokkink, A. H. Westphal, M. Kamperman, J. M. Kleijn and J. W. Borst, Langmuir, 2020, 36, 8494–8502 CrossRef CAS PubMed.
- S. S. Nasr, S. Lee, D. Thiyagarajan, A. Boese, B. Loretz and C.-M. Lehr, Pharmaceutics, 2021, 13, 1924 CrossRef CAS.
- S. S. Nasr, P. Paul, B. Loretz and C.-M. Lehr, Drug Delivery Transl. Res., 2024, 14, 3339–3353 CrossRef CAS.
- E. Ban, M. Park, Y. Kim, J. Park and A. Kim, J. Drug Delivery Sci. Technol., 2024, 97, 105767 CrossRef CAS.
- J. B. Sabadini, C. L. P. Oliveira and W. Loh, Langmuir, 2024, 40, 2015–2027 CrossRef CAS PubMed.
- T.-Y. Heo, D. J. Audus and S.-H. Choi, ACS Macro Lett., 2023, 12, 1396–1402 CrossRef CAS PubMed.
- T.-Y. Heo and S.-H. Choi, J. Phys. Chem. B, 2024, 128, 1256–1265 CrossRef CAS PubMed.
- Y. Zhang, Y. Chen, X. Yang, X. He, M. Li, S. Liu, K. Wang, J. Liu and S. Mann, J. Am. Chem. Soc., 2021, 143, 2866–2874 CrossRef CAS PubMed.
- C.-W. Yeh and Y. Wang, Macromol. Biosci., 2023, 23, 2200538 CrossRef CAS.
- X. Meng, S. L. Perry and J. D. Schiffman, ACS Macro Lett., 2017, 6, 505–511 CrossRef CAS PubMed.
- X. Meng, Y. Du, Y. Liu, E. B. Coughlin, S. L. Perry and J. D. Schiffman, Macromolecules, 2021, 54, 5033–5042 CrossRef CAS.
- F. Weinbreck, R. H. Tromp and C. G. De Kruif, Biomacromolecules, 2004, 5, 1437–1445 CrossRef CAS PubMed.
- J. Zhang, H. Du, N. Ma, L. Zhong, G. Ma, F. Pei, H. Chen and Q. Hu, Food Sci. Hum. Wellness, 2023, 12, 183–191 CrossRef CAS.
- X. Zhou, X. Feng, W. Qi, J. Zhang and L. Chen, Food Hydrocolloids, 2024, 151, 109794 CrossRef CAS.
- H. Liang and J. J. De Pablo, Macromolecules, 2022, 55, 4146–4158 CrossRef CAS.
- F. Akcay Ogur, S. Mamasoglu, S. L. Perry, F. A. Akin and A. B. Kayitmazer, J. Phys. Chem. B, 2024, 128, 9022–9035 CrossRef CAS PubMed.
- C. Love, J. Steinkühler, D. T. Gonzales, N. Yandrapalli, T. Robinson, R. Dimova and T.-Y. D. Tang, Angew. Chem., Int. Ed., 2020, 132, 6006–6013 CrossRef.
- L. Li, B. Lai, J.-N. Yan, M. H. Yambazi, C. Wang and H.-T. Wu, Food Hydrocolloids, 2024, 148, 109445 CrossRef CAS.
- G. Zhang, Y. Wang, P. Cui, Y. Qiu, S. Ye and A. Zhang, Process Biochem., 2024, 143, 292–301 CrossRef CAS.
- T. Lu, K. K. Nakashima and E. Spruijt, J. Phys. Chem. B, 2021, 125, 3080–3091 CrossRef CAS PubMed.
- J. B. Sabadini, C. L. P. Oliveira and W. Loh, J. Colloid Interface Sci., 2025, 678, 1012–1021 CrossRef CAS PubMed.
- X. Liu, Z. Shi, F. Yu, C. Teng, C. Zhang and Z.-R. Chen, Aggregate, 2023, 4, e363 CrossRef CAS.
- F. Späth, A. S. Maier, M. Stasi, A. M. Bergmann, K. Halama, M. Wenisch, B. Rieger and J. Boekhoven, Angew. Chem., Int. Ed., 2023, 62, e202309318 CrossRef PubMed.
- J. Feng, H. Tian, X. Chen, X. Cai, X. Shi and S. Wang, Food Hydrocolloids, 2023, 138, 108439 CrossRef CAS.
- Q. Hu, H. Lan, Y. Tian, X. Li, M. Wang, J. Zhang, Y. Yu, W. Chen, L. Kong, Y. Guo and Z. Zhang, J. Controlled Release, 2024, 365, 176–192 CrossRef CAS PubMed.
- H. Zhang, J. Vandesompele, K. Braeckmans, S. C. D. Smedt and K. Remaut, Chem. Soc. Rev., 2024, 53, 317–360 RSC.
- D. Sun, W. Tan, J. Zhao, Y. Tian, S. Li, Z. Zhang, X. Dong, X. Liu, N. Liu, P. Jiao and J. Ma, Fundam. Res., 2024 DOI:10.1016/j.fmre.2023.11.020.
- S. Chakraborty, Sens. Diagn., 2024, 3, 536–561 RSC.
- Y. B. Yu, K. T. Briggs, M. B. Taraban, R. G. Brinson and J. P. Marino, Pharm. Res., 2021, 38, 3–7 CrossRef CAS PubMed.
- W. C. Blocher and S. L. Perry, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1442 Search PubMed.
- N. Pippa, M. Karayianni, S. Pispas and C. Demetzos, Int. J. Pharm., 2015, 491, 136–143 CrossRef CAS PubMed.
- S. P. Moulik, A. K. Rakshit, A. Pan and B. Naskar, Colloids Interfaces, 2022, 6, 45 CrossRef CAS.
- L.-W. Chang, T. K. Lytle, M. Radhakrishna, J. J. Madinya, J. Vélez, C. E. Sing and S. L. Perry, Nat. Commun., 2017, 8, 1273 CrossRef PubMed.
- Z. Lin, T. Beneyton, J.-C. Baret and N. Martin, Small Methods, 2023, 7, 2300496 CrossRef CAS PubMed.
- J. E. Laaser, Y. Jiang, S. R. Petersen, T. M. Reineke and T. P. Lodge, J. Phys. Chem. B, 2015, 119, 15919–15928 CrossRef CAS PubMed.
- S. Chuanoi, Y. Anraku, M. Hori, A. Kishimura and K. Kataoka, Biomacromolecules, 2014, 15, 2389–2397 CrossRef CAS PubMed.
- J. J. Madinya, H. Tjo, X. Meng, I. A. Ramírez Marrero, C. E. Sing and S. L. Perry, Macromolecules, 2023, 56, 3973–3988 CrossRef CAS.
- I. Benavides, W. A. Scott, X. Cai, Z. H. Zhou and T. J. Deming, Eur. Phys. J. E: Soft Matter Biol. Phys., 2023, 46, 81 CrossRef CAS PubMed.
- A. Hernandez-Garcia, A. H. Velders, M. A. C. Stuart, R. de Vries, J. W. M. van Lent and J. Wang, Chem. – Eur. J., 2017, 23, 239–243 CrossRef CAS PubMed.
- J. Wang, T. Lu, Y. Li, J. Wang and E. Spruijt, Adv. Colloid Interface Sci., 2023, 318, 102964 CrossRef CAS PubMed.
- H. J. Kim, B. Yang, T. Y. Park, S. Lim and H. J. Cha, Soft Matter, 2017, 13, 7704–7716 RSC.
- A. D. Slootbeek, M. H. I. van Haren, I. B. A. Smokers and E. Spruijt, Chem. Commun., 2022, 58, 11183–11200 RSC.
- B. Saini, R. R. Singh, D. Nayak and T. K. Mukherjee, ACS Appl. Nano Mater., 2020, 3, 5826–5837 CrossRef CAS.
- I. Bos, E. Brink, L. Michels and J. Sprakel, Soft Matter, 2022, 18, 2012–2027 RSC.
- J. Wang, W. Guan, T. Tan, V. Saggiomo, M. A. C. Stuart and A. H. Velders, Soft Matter, 2020, 16, 2953–2960 RSC.
- G. Sinani, M. E. Durgun, E. Cevher and Y. Özsoy, Pharmaceutics, 2023, 15, 2021 CrossRef CAS PubMed.
- R. Kembaren, J. M. Kleijn, J. W. Borst, M. Kamperman and A. H. Hofman, Soft Matter, 2022, 18, 3052–3062 RSC.
- Y. Zhang, Y. Huang and S. Li, AAPS PharmSciTech, 2014, 15, 862–871 CrossRef CAS PubMed.
- J. R. Magana, C. C. M. Sproncken and I. K. Voets, Polymers, 2020, 12, 1953 CrossRef CAS PubMed.
- C.-G. Wang, N. E. B. Surat’man, J. J. Chang, Z. L. Ong, B. Li, X. Fan, X. J. Loh and Z. Li, Chem. – Asian J., 2022, 17, e202200604 CrossRef CAS PubMed.
- X. Chen, X. Zhang, S. Yang, J. Wang, T. Tang and M. Gou, Chin. Chem. Lett., 2024, 110021 CrossRef CAS.
- B. Wu, R. W. Lewis, G. Li, Y. Gao, B. Fan, B. Klemm, J. Huang, J. Wang, M. A. C. Stuart and R. Eelkema, Chem. Sci., 2023, 14, 1512–1523 RSC.
- S. Deshpande, F. Brandenburg, A. Lau, M. G. F. Last, W. K. Spoelstra, L. Reese, S. Wunnava, M. Dogterom and C. Dekker, Nat. Commun., 2019, 10, 1800 CrossRef PubMed.
- A. B. Cook, S. Novosedlik and J. C. M. van Hest, Acc. Mater. Res., 2023, 4, 287–298 CrossRef CAS PubMed.
- Y. Bae and K. Kataoka, Adv. Drug Delivery Rev., 2009, 61, 768–784 CrossRef CAS PubMed.
- T. D. Vogelaar, A. E. Agger, J. E. Reseland, D. Linke, H. Jenssen and R. Lund, Biomacromolecules, 2024, 25, 4267–4280 CrossRef CAS PubMed.
- C. Yan and W. Zhang, in Microencapsulation in the Food Industry, ed. A. G. Gaonkar, N. Vasisht, A. R. Khare and R. Sobel, Academic Press, San Diego, 2014, pp. 125–137 Search PubMed.
- F. M. Menger and B. M. Sykes, Langmuir, 1998, 14, 4131–4137 CrossRef CAS.
- K. Xiao, Y. Yang, X. Xu, J. E. S. Szymanowski, Y. Zhou, G. E. Sigmon, P. C. Burns and T. Liu, Inorg. Chem., 2024, 63, 15331–15339 CrossRef CAS PubMed.
- R. Soussi Hachfi, P. Hamon, F. Rousseau, M.-H. Famelart and S. Bouhallab, Foods, 2023, 12, 1040 CrossRef CAS PubMed.
- A. Holkar, S. Gao, K. Villaseñor, M. Lake and S. Srivastava, Soft Matter, 2024, 20, 5060–5070 RSC.
- A. B. Kayitmazer, A. F. Koksal and E. K. Iyilik, Soft Matter, 2015, 11, 8605–8612 RSC.
- G. R. Abraham, A. S. Chaderjian, A. B. N. Nguyen, S. Wilken and O. A. Saleh, Rep. Prog. Phys., 2024, 87, 066601 CrossRef CAS PubMed.
- P. L. Onuchic, A. N. Milin, I. Alshareedah, A. A. Deniz and P. R. Banerjee, Sci. Rep., 2019, 9, 12161 CrossRef PubMed.
- M. Abbas, W. P. Lipiński, J. Wang and E. Spruijt, Chem. Soc. Rev., 2021, 50, 3690–3705 RSC.
- F. Ardestani, A. Haghighi Asl and A. Rafe, Chem. Biol. Technol. Agric., 2022, 9, 83 CrossRef CAS.
- J. Reijenga, A. van Hoof, A. van Loon and B. Teunissen, Anal. Chem. Insights, 2013, 8, ACI.S12304 CrossRef PubMed.
- G. Henriksson, A. Englund, G. Johansson and P. Lundahl, Electrophoresis, 1995, 16, 1377–1380 CrossRef CAS PubMed.
- I. G. Darvey, Biochem. Educ., 1995, 23, 80–82 CrossRef CAS.
- K. Maslowska-Jarzyna, K. M. Bąk, B. Zawada and M. J. Chmielewski, Chem. Sci., 2022, 13, 12374–12381 RSC.
- G. Platzer, M. Okon and L. P. McIntosh, J. Biomol. NMR, 2014, 60, 109–129 CrossRef CAS PubMed.
- R. Lunkad, A. Murmiliuk, Z. Tošner, M. Štěpánek and P. Košovan, Polymers, 2021, 13, 214 CrossRef CAS PubMed.
- S. Pahari, L. Sun and E. Alexov, Database, 2019, 2019, baz024 CrossRef PubMed.
- J. N. Aronson, Biochem. Educ., 1983, 11, 68 CrossRef CAS.
- Y. Tian, Q. Hu, Z. Sun, Y. Yu, X. Li, T. Tian, X. Bi, Y. Li, B. Niu and Z. Zhang, Small, 2024, 20, 2311890 CrossRef CAS PubMed.
- Y. Sun, S. Y. Lau, Z. W. Lim, S. C. Chang, F. Ghadessy, A. Partridge and A. Miserez, Nat. Chem., 2022, 14, 274–283 CrossRef CAS PubMed.
- H. Karoui, M. J. Seck and N. Martin, Chem. Sci., 2021, 12, 2794–2802 RSC.
- J. T. King and A. Shakya, Biophys. J., 2021, 120, 1139–1149 CrossRef CAS PubMed.
- D. Lachowicz, A. Kaczyńska, A. Bodzon-Kulakowska, A. Karewicz, R. Wirecka, M. Szuwarzyński and S. Zapotoczny, Int. J. Mol. Sci., 2020, 21, 9664 CrossRef CAS PubMed.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- N. Deng and W. T. S. Huck, Angew. Chem., Int. Ed., 2017, 56, 9736–9740 CrossRef CAS PubMed.
- D. E. Bergbreiter, A. J. Mijalis and H. Fu, J. Chem. Educ., 2012, 89, 675–677 CrossRef CAS.
- L. Tavares and C. P. Z. Noreña, Food Measure, 2022, 16, 295–306 CrossRef.
- I. Naassaoui and A. Aschi, Eur. Biophys. J., 2021, 50, 877–887 CrossRef CAS PubMed.
- A. Veis and C. Aranyi, J. Phys. Chem., 1960, 64, 1203–1210 CrossRef CAS.
- S. L. Perry, L. Leon, K. Q. Hoffmann, M. J. Kade, D. Priftis, K. A. Black, D. Wong, R. A. Klein, C. F. Pierce, K. O. Margossian, J. K. Whitmer, J. Qin, J. J. De Pablo and M. Tirrell, Nat. Commun., 2015, 6, 6052 CrossRef CAS PubMed.
- Y. Chen and W. Ma, PLoS Comput. Biol., 2020, 16, e1007592 CrossRef CAS PubMed.
- S. F. Ozturk, D. D. Sasselov and J. D. Sutherland, J. Chem. Phys., 2023, 159, 061102 CrossRef CAS PubMed.
- M. G. Weller, Life, 2024, 14, 341 CrossRef CAS PubMed.
- G. T. M. Bitchagno, V.-A. Nchiozem-Ngnitedem, D. Melchert and S. A. Fobofou, Nat. Rev. Chem., 2022, 6, 806–822 CrossRef PubMed.
- K. M. Lebold and R. B. Best, J. Phys. Chem. B, 2022, 126, 2407–2419 CrossRef CAS PubMed.
- Y. H. Kim, K. Lee and S. Li, Chem. Mater., 2021, 33, 7923–7943 CrossRef CAS.
- Y. Wang, R. Zou, Y. Zhou, Y. Zheng, C. Peng, Y. Liu, H. Tan, Q. Fu and M. Ding, Chem. Sci., 2024, 15, 13442–13451 RSC.
- E. A. Frankel, P. C. Bevilacqua and C. D. Keating, Langmuir, 2016, 32, 2041–2049 CrossRef CAS PubMed.
- D. Priftis, X. Xia, K. O. Margossian, S. L. Perry, L. Leon, J. Qin, J. J. De Pablo and M. Tirrell, Macromolecules, 2014, 47, 3076–3085 CrossRef CAS.
- T. Lin, Y. Zhou, Y. Dadmohammadi, M. Yaghoobi, G. Meletharayil, R. Kapoor and A. Abbaspourrad, Food Hydrocolloids, 2023, 145, 109064 CrossRef CAS PubMed.
- J. Zhou, Y. Cai, Y. Wan, B. Wu, J. Liu, X. Zhang, W. Hu, M. A. Cohen Stuart and J. Wang, J. Colloid Interface Sci., 2023, 650, 2065–2074 CrossRef CAS PubMed.
- W. C. Blocher McTigue and S. L. Perry, Soft Matter, 2019, 15, 3089–3103 RSC.
- W. C. Blocher McTigue and S. L. Perry, Small, 2020, 16, 1907671 CrossRef CAS PubMed.
- R. G. Ingle and W.-J. Fang, Pharmaceutics, 2023, 15, 1158 CrossRef CAS PubMed.
- K. Yadav, K. K. Sahu, Sucheta, S. P. E. Gnanakani, P. Sure, R. Vijayalakshmi, V. D. Sundar, V. Sharma, R. Antil, M. Jha, S. Minz, A. Bagchi and M. Pradhan, Int. J. Biol. Macromol., 2023, 241, 124582 CrossRef CAS PubMed.
- L. Zhu, J. Luo and K. Ren, J. Mater. Chem. B, 2023, 11, 261–279 RSC.
- M. Lu, H. Xing, A. Zheng, Y. Huang and X.-J. Liang, Acc. Chem. Res., 2023, 56, 224–236 CrossRef CAS PubMed.
- R. J. Mosley, B. Rucci and M. E. Byrne, J. Mater. Chem. B, 2023, 11, 2078–2094 RSC.
- Z.-G. Lu, J. Shen, J. Yang, J.-W. Wang, R.-C. Zhao, T.-L. Zhang, J. Guo and X. Zhang, Signal Transduction Targeted Ther., 2023, 8, 1–53 CrossRef CAS PubMed.
- J. R. Vieregg, M. Lueckheide, A. B. Marciel, L. Leon, A. J. Bologna, J. R. Rivera and M. V. Tirrell, J. Am. Chem. Soc., 2018, 140, 1632–1638 CrossRef CAS PubMed.
- C. Forenzo and J. Larsen, Mol. Pharmaceutics, 2023, 20, 4387–4403 CrossRef CAS PubMed.
- S. M. Naghib, B. Ahmadi, B. Mikaeeli Kangarshahi and M. R. Mozafari, Int. J. Biol. Macromol., 2024, 278, 134542 CrossRef CAS PubMed.
- L. Ma, X. Fang and C. Wang, Front. Bioeng. Biotechnol., 2023, 10, 1100365 CrossRef PubMed.
- Z. Wang, X. Zhang, M. Han, X. Jiao, J. Zhou, X. Wang, R. Su, Y. Wang and W. Qi, J. Mater. Chem. B, 2023, 11, 8974–8984 RSC.
- Y. Kakizawa and K. Kataoka, Adv. Drug Delivery Rev., 2002, 54, 203–222 CrossRef CAS PubMed.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- H. Cho, J. Kim, S. Kim, Y. C. Jung, Y. Wang, B.-J. Kang and K. Kim, J. Controlled Release, 2020, 327, 284–295 CrossRef CAS PubMed.
- H. J. Kim, A. Kim, K. Miyata and K. Kataoka, Adv. Drug Delivery Rev., 2016, 104, 61–77 CrossRef CAS PubMed.
- S. Abbasi, S. Uchida, K. Toh, T. A. Tockary, A. Dirisala, K. Hayashi, S. Fukushima and K. Kataoka, J. Controlled Release, 2021, 332, 260–268 CrossRef CAS PubMed.
- R. Aman, M. M. Syed, A. Saleh, F. Melliti, S. R. Gundra, Q. Wang, T. Marsic, A. Mahas and M. M. Mahfouz, Nucleic Acids Res., 2024, gkae148 Search PubMed.
- R. A. Kapelner, V. Yeong and A. C. Obermeyer, Curr. Opin. Colloid Interface Sci., 2021, 52, 101407 CrossRef CAS.
- P. Kaushik, P. K. Pandey, V. K. Aswal and H. B. Bohidar, Soft Matter, 2020, 16, 9525–9533 RSC.
- X. Zhang, Y. Lin, N. A. Eschmann, H. Zhou, J. N. Rauch, I. Hernandez, E. Guzman, K. S. Kosik and S. Han, PLoS Biol., 2017, 15, e2002183 CrossRef PubMed.
- P. Chowdhury, B. Saha, K. Bauri, B. S. Sumerlin and P. De, J. Am. Chem. Soc., 2024, 146, 21664–21676 CrossRef PubMed.
- T. Lu, S. Javed, C. Bonfio and E. Spruijt, Small Methods, 2023, 7, 2300294 CrossRef CAS PubMed.
- A.-G. Niculescu, A. C. Bîrcă and A. M. Grumezescu, Pharmaceutics, 2021, 13, 2053 CrossRef CAS PubMed.
- P. U. Joshi, C. Decker, X. Zeng, A. Sathyavageeswaran, S. L. Perry and C. L. Heldt, Biomacromolecules, 2024, 25(2), 741–753, DOI:10.1021/acs.biomac.3c00938.
- T. Sneideris, N. A. Erkamp, H. Ausserwöger, K. L. Saar, T. J. Welsh, D. Qian, K. Katsuya-Gaviria, M. L. L. Y. Johncock, G. Krainer, A. Borodavka and T. P. J. Knowles, Nat. Commun., 2023, 14, 7170 CrossRef CAS PubMed.
- M. Peydayesh, S. Kistler, J. Zhou, V. Lutz-Bueno, F. D. Victorelli, A. B. Meneguin, L. Spósito, T. M. Bauab, M. Chorilli and R. Mezzenga, Nat. Commun., 2023, 14, 1848 CrossRef CAS PubMed.
- K. Roy, H.-Q. Mao, S.-K. Huang and K. W. Leong, Nat. Med., 1999, 5, 387–391 CrossRef CAS PubMed.
- H. Nishida, Y. Matsumoto, K. Kawana, R. J. Christie, M. Naito, B. S. Kim, K. Toh, H. S. Min, Y. Yi, Y. Matsumoto, H. J. Kim, K. Miyata, A. Taguchi, K. Tomio, A. Yamashita, T. Inoue, H. Nakamura, A. Fujimoto, M. Sato, M. Yoshida, K. Adachi, T. Arimoto, O. Wada-Hiraike, K. Oda, T. Nagamatsu, N. Nishiyama, K. Kataoka, Y. Osuga and T. Fujii, J. Controlled Release, 2016, 231, 29–37 CrossRef CAS PubMed.
- D. Zhou, C. Li, Y. Hu, H. Zhou, J. Chen, Z. Zhang and T. Guo, J. Mater. Chem., 2012, 22, 10743–10751 RSC.
- A. Herrmann, S. Abbina, N. V. Bathula, P. M. M. Nouri, I. Chafeeva, I. Constantinescu, E. Abbasi, U. Abbasi, M. Drayton, H. D. Luo, H. Moon, A. Gill, Y. Xi, A. K. Bertram, C. Du, R. Haag, A. K. Blakney and J. N. Kizhakkedathu, Adv. Funct. Mater., 2024, 34, 2406878 CrossRef CAS.
- W. Liu, J. Deng, S. Song, S. Sethi and A. Walther, Commun. Chem., 2024, 7, 1–10 CrossRef PubMed.
- C. Delehedde, I. Ciganek, N. Rameix, N. Laroui, C. Gonçalves, L. Even, P. Midoux and C. Pichon, Int. J. Pharm., 2023, 647, 123531 CrossRef CAS PubMed.
- L. Mott, C. Akers and D. W. Pack, J. Drug Delivery Sci. Technol., 2023, 84, 104465 CrossRef CAS.
- C. Salvador, P. Andreozzi, G. Romero, I. Loinaz, D. Dupin and S. E. Moya, ACS Appl. Bio Mater., 2023, 6, 529–542 CrossRef CAS PubMed.
- Y. Li, B. Humphries, Z. Wang, S. Lang, X. Huang, H. Xiao, Y. Jiang and C. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 30735–30746 CrossRef CAS PubMed.
- Y. Cai, N. Y. Naser, J. Ma and F. Baneyx, Biomacromolecules, 2024, 25, 2390–2398 CrossRef CAS PubMed.
- K. Yoshida, K. Suyama, S. Matsushita, I. Maeda and T. Nose, J. Pept. Sci., 2023, 29, e3528 CrossRef CAS PubMed.
- R. Leicher, A. Osunsade, G. N. L. Chua, S. C. Faulkner, A. P. Latham, J. W. Watters, T. Nguyen, E. C. Beckwitt, S. Christodoulou-Rubalcava, P. G. Young, B. Zhang, Y. David and S. Liu, Nat. Struct. Mol. Biol., 2022, 29, 463–471 CrossRef CAS PubMed.
- Y. Zhou, K. Zhang, S. Moreno, A. Temme, B. Voit and D. Appelhans, Angew. Chem., Int. Ed., 2024, 63, e202407472 CrossRef CAS PubMed.
- S. Cao, T. Ivanov, J. Heuer, C. T. J. Ferguson, K. Landfester and L. Caire Da Silva, Nat. Commun., 2024, 15, 39 CrossRef CAS PubMed.
- L. Zhang, M. Chen, Z. Wang, M. Zhong, H. Chen, T. Li, L. Wang, Z. Zhao, X.-B. Zhang, G. Ke, Y. Liu and W. Tan, ACS Cent. Sci., 2024, 10, 1201–1210 CrossRef CAS PubMed.
- W. M. Aumiller, F. Pir Cakmak, B. W. Davis and C. D. Keating, Langmuir, 2016, 32, 10042–10053 CrossRef CAS PubMed.
- A. I. Bhat and G. P. Rao, in Characterization of Plant Viruses: Methods and Protocols, ed. A. I. Bhat and G. P. Rao, Springer, US, New York, NY, 2020, pp. 377–381 Search PubMed.
- R. K. Kumar, R. L. Harniman, A. J. Patil and S. Mann, Chem. Sci., 2016, 7, 5879–5887 RSC.
- S. Gao and S. Srivastava, ACS Macro Lett., 2022, 11, 902–909 CrossRef CAS PubMed.
- C. E. R. Edwards, K. L. Lakkis, Y. Luo and M. E. Helgeson, Soft Matter, 2023, 19, 8849–8862 RSC.
- V. Lauth, M. Maas and K. Rezwan, J. Mater. Chem. B, 2014, 2, 7725–7731 RSC.
- K. K. Nakashima, A. A. M. André and E. Spruijt, in Methods in Enzymology, ed. C. D. Keating, Academic Press, 2021, vol. 646, pp. 353–389 Search PubMed.
- N. S. Heredia, K. Vizuete, M. Flores-Calero, V. K. Pazmiño, F. Pilaquinga, B. Kumar and A. Debut, PLoS One, 2022, 17, e0264825 CrossRef CAS PubMed.
- F. Ardestani, A. Haghighi Asl and A. Rafe, Chem. Biol. Technol. Agric., 2024, 11, 118 CrossRef CAS.
- A. Salehi, A. Rezaei, M. S. Damavandi, M. S. Kharazmi and S. M. Jafari, Food Chem., 2023, 405, 134801 CrossRef CAS PubMed.
- M. H. M. E. van Stevendaal, L. Vasiukas, N. A. Yewdall, A. F. Mason and J. C. M. van Hest, ACS Appl. Mater. Interfaces, 2021, 13, 7879–7889 CrossRef CAS PubMed.
- M. H. van Haren, K. K. Nakashima and E. Spruijt, J. Syst. Chem., 2020, 8, 107–120 Search PubMed.
- D. L. Rohlfing, Origins Life Evol. Biospheres, 1975, 6, 203–209 CrossRef CAS PubMed.
- X. Mi, W. C. Blocher McTigue, P. U. Joshi, M. K. Bunker, C. L. Heldt and S. L. Perry, Biomater. Sci., 2020, 8, 7082–7092 RSC.
- H. Chen, Y. Bao, X. Li, F. Chen, R. Sugimura, X. Zeng and J. Xia, Angew. Chem., Int. Ed., 2024, e202410566 CAS.
- J. Li and K. Kataoka, J. Am. Chem. Soc., 2021, 143, 538–559 CrossRef CAS PubMed.
- K. Koji, N. Yoshinaga, Y. Mochida, T. Hong, T. Miyazaki, K. Kataoka, K. Osada, H. Cabral and S. Uchida, Biomaterials, 2020, 261, 120332 CrossRef CAS PubMed.
- H. Cabral, H. Kinoh and K. Kataoka, Acc. Chem. Res., 2020, 53, 2765–2776 CrossRef CAS PubMed.
- S. Yousefi Adlsadabad, J. W. Hanrahan and A. Kakkar, Int. J. Mater. Sci., 2024, 25, 1739 CAS.
- M. Lin, X. Lv, H. Wang, L. Shu, H. Wang, G. Zhang, J. Sun and X. Chen, Adv. Mater., 2024, 36, 2407378 CrossRef CAS PubMed.
- X. Li, E. K. Dowling, G. Yan, Z. Dereli, B. Bozorgui, P. Imanirad, J. H. Elnaggar, A. Luna, D. G. Menter, P. G. Pilié, T. A. Yap, S. Kopetz, C. Sander and A. Korkut, Cancer Discovery, 2022, 12, 1542–1559 CrossRef CAS PubMed.
- S. K. Yadava, B. P. K. Reddy, M. R. Prausnitz and M. T. Cicerone, ACS Appl. Mater. Interfaces, 2024, 16, 15981–15992 CrossRef CAS PubMed.
- R. Tenchov, J. M. Sasso and Q. A. Zhou, Bioconjugate Chem., 2023, 34, 941–960 CrossRef CAS PubMed.
- Y. Liu, Y. Huang, G. He, C. Guo, J. Dong and L. Wu, Int. J. Mol. Sci., 2024, 25, 10166 CrossRef CAS PubMed.
- F. Pir Cakmak, A. T. Grigas and C. D. Keating, Langmuir, 2019, 35, 7830–7840 CrossRef CAS PubMed.
- Y. Qin, L. Ou, L. Zha, Y. Zeng and L. Li, Mol. Biomed., 2023, 4, 48 CrossRef CAS PubMed.
- Y. Xu, T. Fourniols, Y. Labrak, V. Préat, A. Beloqui and A. des Rieux, ACS Nano, 2022, 16, 7168–7196 CrossRef CAS PubMed.
- R. Kaup, J. B. ten Hove, A. Bunschoten, F. W. B. van Leeuwen and A. H. Velders, Nanoscale, 2021, 13, 15422–15430 RSC.
- A. Pica, I. Russo Krauss, V. Parente, H. Tateishi-Karimata, S. Nagatoishi, K. Tsumoto, N. Sugimoto and F. Sica, Nucleic Acids Res., 2017, 45, 461–469 CrossRef CAS PubMed.
- E. Kluczka, V. Rinaldo, A. Coutable-Pennarun, C. Stines-Chaumeil, J. L. R. Anderson and N. Martin, ChemCatChem, 2024, 16, e202400558 CrossRef CAS.
- I. B. A. Smokers, B. S. Visser, A. D. Slootbeek, W. T. S. Huck and E. Spruijt, Acc. Chem. Res., 2024, 57, 1885–1895 CrossRef CAS PubMed.
- U. Odunze, N. Rustogi, P. Devine, L. Miller, S. Pereira, S. Vashist, H. J. Snijder, D. Corkill, A. Sabirsh, J. Douthwaite, N. Bond and A. Desai, Nucleic Acids Res., 2024, 52, 9384–9396 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |