
Comprehensive insights into hydrolysis-mediated hydrogen production using high-entropy quintuple alloy-grafted carbon black†
Gokul
Raj
 a,
Ravi
Nandan‡
a,
Soumen
Midya‡
a,
Kanhai
Kumar
a,
Abhishek Kumar
Singh
a and
Karuna Kar
Nanda
*abc
a,
Ravi
Nandan‡
a,
Soumen
Midya‡
a,
Kanhai
Kumar
a,
Abhishek Kumar
Singh
a and
Karuna Kar
Nanda
*abc
aMaterials Research Centre, Indian Institute of Science, Bangalore-560012, India. E-mail: nanda@iisc.ac.in
bInstitute of Physics, Bhubaneshwar-751005, India
cHomi Bhabha National Institute, Anushaktinagar, Mumbai-400094, India
First published on 27th January 2025
Abstract
Hydrogen is a promising fuel with adequate energy density and zero carbon emissions upon combustion. Its fast and convenient production from hydrogen-rich materials like sodium borohydride (NaBH4) is of high practical importance. Herein, we report a novel quintuple high-entropy alloy of CrMnFeCoNi loaded on conductive carbon (HEACB) as an effective heterogeneous catalyst for the hydrolysis of NaBH4 to produce hydrogen at room temperature. The optimized HEACB shows excellent performance with a turn over freequency (TOF) of about 188 mL gMn−1 s−1 and a minimum activation energy (Ea) of about 15 kJ mol−1, while the Ea of bare HEA is 43 kJ mol−1. Mott–Schottky analysis reveals n-type semiconductor behavior of the HEACB heterostructure at the electrolyte interface during the reaction with an optimal flat band potential. Electrochemical analysis is employed to understand the kinetic aspects of the observed catalytic behavior during hydrolysis. The onset potential, overpotential, Tafel slope, and charge transfer resistance extracted from linear sweep voltammetry and electrochemical impedance studies reveal that the HEA alloy stabilization greatly affects the overall catalytic efficiency towards NaBH4 hydrolysis. Chemical and electrochemical investigations exhibit a pronounced correlation. Density Functional Theory (DFT) is applied to understand the interaction of the reaction intermediates and to provide insights into the hydrolysis process of borohydride on the HEA system.
1. Introduction
Hydrogen has always been a focus point of the energy sector because of its clean, sustainable, and environmentally friendly combustion with zero carbon emissions.1 Moreover, it possesses a high theoretical energy density (120 MJ kg−1), almost three times that of gasoline and diesel. In terms of usable electrical energy, the energy density of hydrogen is equivalent to 33.6 kW h, whereas diesel accounts for only 14 kW h.2 These statistics indicate the importance of the detailed exploration of chemical hydrogen storage materials such as metal boron hydrides, formic acid (HCOOH), and hydrazine (N2H4·H2O) because of their simplicity and safety in transportation, in addition to the ease of generating hydrogen upon their chemical breakdown employing hydrolysis.3 Among these potentially viable hydrogen-dense materials, sodium borohydride has gained considerable attention due to its relatively high hydrogen storage capacity of about 11 percent by weight, non-inflammable nature, safer chemical attributes, and high hydrogen-producing capacity through aqueous hydrolysis.4 In recent times, efforts have been made to utilize this rich source of hydrogen in the alkaline medium-based hydrolysis of NaBH4.5,6 Various heterogeneous catalysts based on precious metals like Pt, Pd, and Ru, along with the transition metal counterparts based on Co, Ni, and Fe have been reported as efficient catalysts that can mediate the hydrolysis of NaBH4.7,8 However for the noble metals, their limited resources, high production cost and poor operational stability limit their practical applicability towards hydrolysis-based fuel production.8In the quest for cost-effective and efficient catalysts, various bimetal alloys of transition metals and noble metals like Ni–Co, Pd–Ni, Pt–Ag, Ru–Cu, Cu–Pt, Co–Fe, Ru2Pt, and Ni2Pt/Fe3O4 have been efficaciously developed recently for the hydrolysis of NaBH4.1,8–11 For better electrochemical surface area utilization and uniform active center distribution, metal–organic framework (MOF)-based composites have also been studied for alkaline NaBH4 hydrolysis.12 To further enhance the catalytic performance and improve the chemical and mechanical stability of such catalytic materials to withstand harsh reaction conditions, various heterostructures based on graphene, carbon nanotubes, MXenes, etc., have been proposed.13–15
As catalytic activity can be improved by alloying with a greater number of metals, we have employed a novel quintuple-component-based high entropy alloy (HEA) for efficient hydrolysis of NaBH4. We have utilized a single-step precursor pyrolysis strategy to stabilize the FCC HEA of Cr, Mn, Fe, Co, and Ni and in situ decorate it on a conductive carbon support. HEAs generally adopt the phase common to the constituent elements because of the extended degree of solubility.16 The other factors like elemental size, electronegativity, and valence electron concentration of the system are also crucial for the stability of the final fcc phase of the HEAs.17–20 Our goal in designing such an FCC HEA is to minimize the use of precious metals like Pt, Ru, and Ir, which suffer from poor operational stability. Furthermore, recent experimental and theoretical studies suggest that NaBH4 hydrolysis needs simultaneous adsorption of borohydride ions and water molecules to release hydrogen.21 HEA offers multiple active sites on a single platform which makes it more feasible. Borohydride adsorption and B–H bond breaking are favoured by an electron-rich active site, which makes the B–H cleavage easier.22 Recently, a few transition metal borides (Co, Fe, and Mn) have been developed for effective catalysis of such hydrolysis by electronic optimization of the metal.23,24 To minimize the toxicity of borides, we attempted to synergistically perturb Co, Mn, and Fe by forming a HEA alloy within 3d-row elements like Cr and Ni. This results in complex synergistic interactions among these five elements, and based on electronegativity and charge density, electrons diffuse from one atomic site to another, which helps to optimize metal centers for better intermediate interaction.25 The synthesized HEA showed promising hydrolysis of NaBH4 with a TOF of about 188 mL gMn−1 s−1 and excellent durability. The activation energy of the reaction is found to be 15 kJ mol−1 from the temperature-dependent hydrogen production kinetics. An electrochemical study was conducted to probe details regarding the catalysis of hydrolysis. To unveil the behavior of the heterogenous catalyst towards the reactive medium, we employed a novel Mott–Schottky approach, which revealed an n-type behavior with optimal band bending at the interface. A poisoning test conducted using cyanide ions revealed that the HEA metal centers are the active sites responsible for the catalysis. DFT calculations were employed to understand the interaction between the intermediates and the active metal center in the alloy system as well as to compare the activity of the HEA carbon composite and bare HEA NPs. The adsorption energy calculation of the intermediate could shed light on the energy barrier of the reaction and the efficiency of the HEA composite in minimizing it during catalysis.
2. Experimental section
2.1. Materials and reagents
Manganese(II) nitrate hexahydrate (Mn(NO3)2·6H2O, Mw = 287.04 g mol−1), cobalt(II) nitrate hexahydrate (Co(NO3)·6H2O, Mw = 291.03 g mol−1), nickel(II) nitrate hexahydrate (Ni(NO3)·6H2O), iron(III) nitrate hexahydrate (Fe(NO3)3·6H2O, Mw = 349.9 g mol−1), chromium nitrate hexahydrate (Cr(NO3)3·6H2O, Mw = 346.10 g mol−1), manganese(III) acetylacetonate Mn(C5H7O2)3, Mw = 352.26 g mol−1, cobalt(II) acetylacetonate, Co(C5H7O2)2, Mw = 257.15 g mol−1, nickel(II) acetylacetonate Ni(C5H7O2)2, Mw = 256.9 g mol−1, iron(III) acetylacetonate Fe(C5H7O2)3, Mw = 353.2 g mol−1, chromium(III) acetylacetonate Cr(C5H7O2)3, Mw = 349.3 g mol−1, carbon black (CAS number: 1333-86-4) were purchased from Sigma Aldrich and used after a specific purification process, along with hydrofluoric acid (35%), and sodium borohydride (99%).2.2. Synthesis route for the high entropy alloy grafted on conductive carbon black (HEACB)
A 0.5 M aqueous solution each of Cr(NO3)3·6H2O, Mn(NO3)2·6H2O, Fe(NO3)3·6H2O, Co(NO3)2·6H2O, and Ni(NO3)2·6H2O was separately prepared and combined under continuous stirring using a magnetic stirrer. The mixture was heated to about 50 °C to improve precursor miscibility. After 2 h of heating on a magnetic hot plate, approximately 2 g of pure carbon black (CB) and 15 mL of ethanol were added to the solution. Following this heat treatment, the mixture was freeze-dried to produce a fluffy dark brown precursor powder. This powder was then subjected to high-temperature pyrolysis at 1000 °C with a ramping rate of 10°C min−1 for 2 h in an inert atmosphere using a tube furnace with a constant nitrogen gas flow of about 100 sccm (Fig. 1), similar to our previous report.26 Three independent samples were fabricated at different synthesis temperatures of 500 °C, 750 °C, and 1000 °C and labeled as HEACB 500, HEACB 750, and HEACB 1000, respectively. To compare the activity of the heterostructure HEACB, bare HEA nanoparticles without carbon support were synthesized separately by employing a solvothermal strategy using an ethanol–acetone mixture.27 Corresponding metal acetylacetonate salts were dissolved in an equimolar concentration (0.5 mM) in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ethanol acetone mixture and heated in an autoclave for 12 h at 200 °C. The supernatant after centrifugation was decanted, washed with acetone, and dried at room temperature. The samples thus prepared are safely stored in a desiccator, and utilized for further analysis.
:50 ethanol acetone mixture and heated in an autoclave for 12 h at 200 °C. The supernatant after centrifugation was decanted, washed with acetone, and dried at room temperature. The samples thus prepared are safely stored in a desiccator, and utilized for further analysis.
 | ||
| Fig. 1 Schematic illustration of the synthesis protocol. | ||
2.3. Characterization
X-ray diffraction (XRD) studies were conducted on a PANalytical branded diffractometer with monochromatized Cu Kα radiation at 1.54 Å wavelength, scanning electron microscopy (SEM) was performed utilizing a high-resolution field-emission microscope (FESEM FEI In-spect 50) and transmission electron microscopy (TEM) was conducted using a high-resolution TEM, Tecnai T20 operating at about 200 kV. XPS was performed on an AXIS ULTRA DLD Kratos instrument with a monochromatic Al Kα radiation source at 1486.6 eV energy.About 50 mg of our HEACB catalyst was homogeneously dispersed in 100 mL of water and maintained under constant agitation using a magnetic stirrer during the NaBH4 hydrolysis experiment. About 0.1 M NaBH4 solution was dispersed onto the heterogenous catalyst mixture to examine the hydrolysis reaction where the pH of the medium was kept neutral. An inverted burette method was used to measure the released hydrogen gas during the hydrolysis of NaBH4. The turnover frequency (TOF) and reaction rate during catalysis were estimated using the quantified hydrogen gas. An Arrhenius temperature-dependent rate study was also performed to determine the effective activation barrier involved during the catalysis of the reaction. A poisoning test using sodium cyanide was used to evaluate the involvement of metal active centers on our heterogenous catalyst HEA PB towards the hydrolysis of NaBH4 as per the reaction shown in eqn (1).
| NaBH4(s) + H2O(l) → NaB(OH)4(aq) + H2(g) | (1) |
To gain an electrochemical perspective on the NaBH4 oxidation process, including hydrogen evolution, we conducted a thorough electro-oxidation of the NaBH4 substrate and assessed multiple kinetic features of the reaction. The electrochemical investigations were conducted using a CHI 750E apparatus with a three-electrode configuration. A commercial glassy carbon electrode (GCE) with a surface area of 0.07 cm2 was employed. The reference electrode in this experiment was Ag/AgCl, while the counter electrode was made of graphite. Approximately 7 mg of catalyst material was added to a 1000 μL ethanol–water solution for each electrochemical analysis. A 5 wt% Nafion 117 solution (20 μL) was added to the produced mixture (100 μL) to create a well-dispersed thick catalyst ink. The mixture was then sonicated for 15 minutes. Afterward, 7 μL of this catalyst ink was drop-cast onto the electrode for each electrochemical assessment. The NaBH4 oxidation measurements were conducted in a 1 M KOH electrolyte solution at a scan rate of 10 mV s−1.
2.4. Computational methodology
All spin-polarized Density Functional Theory (DFT) calculations were carried out using the Vienna Ab initio Simulation Package (VASP).28 To describe electron–ion interactions all electron-projected augmented wave pseudopotentials29 were used and the exchange correlation functional was approximated using Perdew–Bruke–Ernzerhof (PBE) under the generalized gradient approximation (GGA).30 The Brillouin zone was sampled using a gamma center k points mesh of 15 × 15 × 1 for the graphene unit cell and 2 × 2 × 1 for the 7 × 7 graphene supercell, the amorphous graphene, and the composite heterostructure. The kinetic energy cut-off for the plane wave basis set was set to 500 eV for all the structures. A conjugate gradient scheme was employed for all structural optimizations until the energies were less than 10−5 eV for all the structures and each force component was less than 0.005 eV Å−1 for the graphene unit cell and 0.01 eV Å−1 for all other structures. The Grimme-D3 correction was used to account for the van der Waals (vdW) interaction between the amorphous graphene layer and the HEA nanoparticles.31The formation energy (Ef) of composite heterostructure formation is calculated as follows:
| Ef = Etotal − Edefected_carbon − Enano | (2) |
The adsorption energy (ΔEads) of various intermediates is calculated using the following formula,
| ΔEads = Etotal − Ecatalyst − Eintermediate | (3) |
3. Results and discussion
3.1. Characterization details of HEACBs
A temperature-dependent phase stabilization study of the samples was performed using XRD as shown in Fig. 2a and S1.† HEACBs prepared at three different temperatures of 500, 750 and 1000 °C were analyzed for their crystal structure. It was observed that the sample prepared at 500 °C exhibited peaks corresponding to the BCC phase of the HEA alloy centered at 43.4°, and 63°. A metal carbide peak at around 37.4° was also observed, indicating partial reduction at this temperature.33–35 On raising the temperature to about 750 °C, three prominent peaks centered at 44.3°, 51.5°, and 75.4° appeared corresponding to the FCC phase of HEA alloys and the metal carbide peak disappeared indicating the complete reduction of all metals.34,35 Peaks around 30.2°, and 75.7° indicate the formation of a B2 phase in the system.36,37 A new peak around 35.4° appeared at this temperature indicating the formation of a Laves phase.38,39 On raising the synthesis temperature further to 1000 °C, the intensification of FCC peaks was observed and the BCC peaks were diminished. The Laves peak around 35.4° was found to intensify slightly at this temperature along with the appearance of a diminished HCP peak around 56.9° indicating that raising the temperature above 1000 °C can result in the appearance of new phases and a loss of phase purity of HEAs. However, the higher intensity of the FCC phase shows that 1000 °C is the optimal temperature for stabilization of the FCC-phased HEA alloy on the conductive carbon support. The lattice constant, strain and crystallite size of the HEACBs were estimated and are detailed in Table S1.† We observed that on raising the synthesis temperature, the crystallite size also increases.40 Raman spectroscopic analysis in Fig. 2b shows the presence of defective carbon support with a prominent defective peak centered around 1350 cm−1 (D band) and its overtone at around 2690 cm−1.41 The overtone position is dependent on the incident laser energy.42 The defects in the carbon matrix on which our HEA nanoparticles are uniformly grafted create a better environment for the adsorption of intermediates during the reaction and favor the catalytic activity.43–45 The ID/IG ratio of HEACB 1000 is 0.99, while the ratio is merely about 0.87 for HEACB 500. On comparing the defects to the graphitic ratio, it seems that higher temperatures induce the formation of a higher degree of defects in the heterostructure.46–48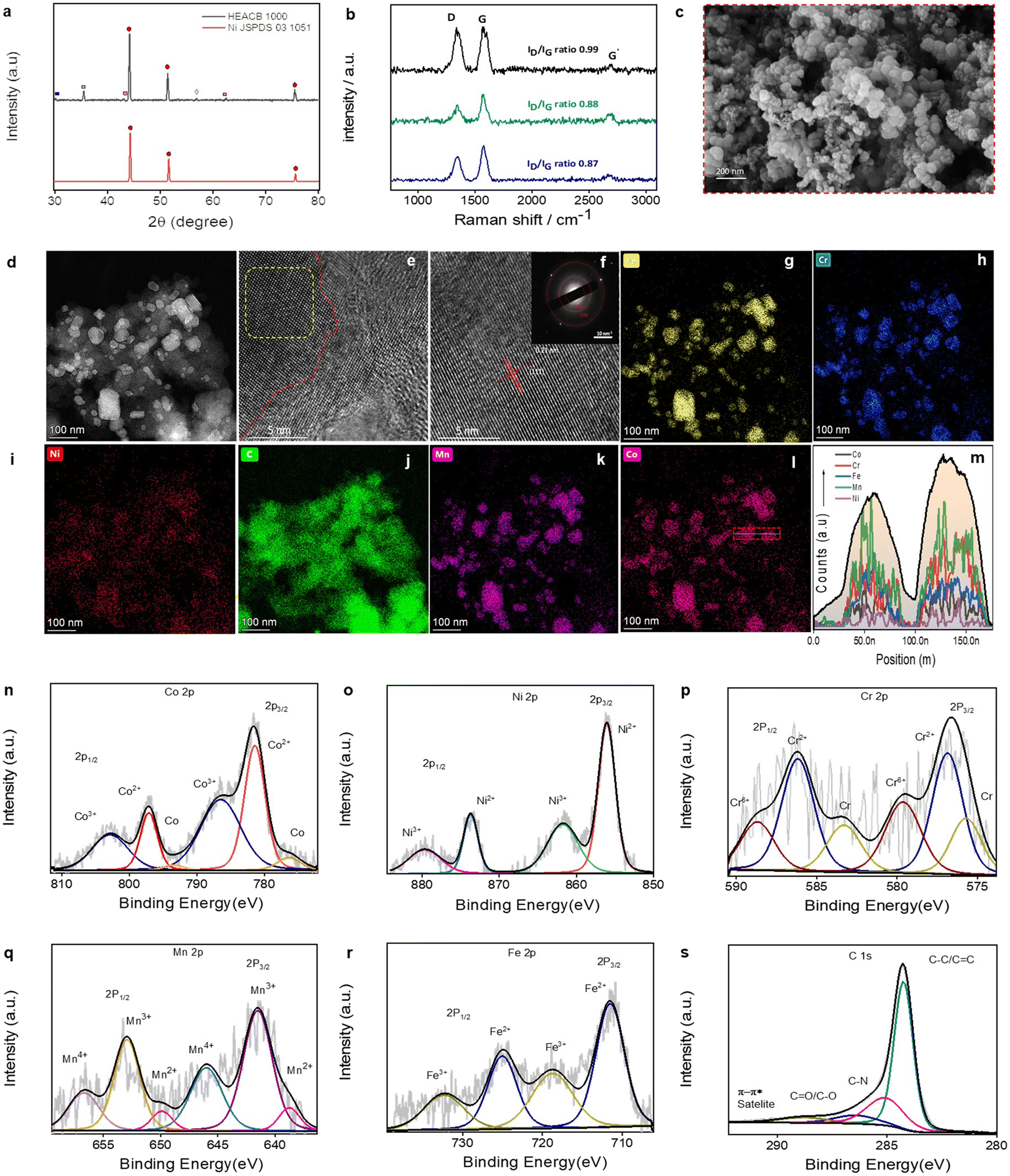 | ||
| Fig. 2 Structural and chemical characterization of the HEACB heterostructure: (a) XRD pattern of HEACB 1000 in comparison to Ni FCC, (b) Raman spectra of HEACBs comparing their defect ratio, (c) SEM image of HEACB 1000, (d) HAADF dark field image of HEACB 1000, (e and f) HR TEM image of HEACB 1000 (inset: SAED pattern), (g–l) elemental mapping on HEACB 1000 of a representative region in HAADF STEM mode, (m) EDS line scan over two representative HEACB 1000 nanoparticles, and (n–s) resolved high resolution XPS spectra of Co 2p, Ni 2p, Cr 2p, Mn 2p, Fe 2p, and C 1s, respectively. | ||
The preliminary morphological examination of the HEACB 1000 heterostructure was done using SEM and the image is shown in Fig. 2c and S2.† No considerable change was observed in the overall morphology of the heterostructure concerning the synthesis temperature. Fig. S3† shows the Energy Dispersive X-ray Spectroscopy (EDS) elemental mapping of HEACB 1000 for a representative region. The five metals, Cr to Ni, in the 3d transition series were found to be uniformly distributed onto the observed particles without phase segregation. The relative atomic percentage of each metal extracted from the EDS elemental spectrum is listed in Table S2.† The ratios of the constituent metals Cr:Mn:Fe:Co:Ni were found to be 23.3:33:16.5:13.6:13.6. The entropy, calculated based on the relative composition, is ∼1.54R, which is well within the prescribed range for HEA classification. The ICPMS data analysis (Table S3†) of elements also supports this finding. The dark-field TEM images as shown in Fig. 2d indicate the formation of nanoparticles grafted on the carbon support. The HRTEM images in Fig. 2e and f show the lattice fringes of the HEA alloys. The atomic contrast seen in Fig. S4† represents the unit cell with different atoms, indicative of successful alloying. The particle size distribution analysis from the TEM images shown in Fig. S5† indicated an average particle size of about 64 nm. The comparative TEM analysis of HEACB and bare HEA shown in Fig. S6† reveals that CB plays a considerable role in the uniform distribution of HEA particles. In the case of HEACBs, the alloy nanoparticles are distributed over the entire carbon support as discrete particles with less agglomeration. On the other hand, we can see that the particles are agglomerated at many places in the case of bare HEAs. However, the particle size of HEAs in the presence of the carbon support is found to be larger compared to bare HEAs. This is because of the faster and feasible reduction of HEA metals in the presence of carbon at elevated temperatures, leading to swift nucleation and growth.49,50 The EDS elemental mapping of a few representative nanoparticles is shown in Fig. 2g–l, with a specific EDS line scan over two randomly selected HEA nanoparticles presented in Fig. 2m, ensuring the homogeneous distribution of elements at the nanoparticle level. The elemental composition was also determined through inductively coupled plasma mass spectroscopy (ICPMS) and XPS measurements (Tables S3 and S4†). The intrinsic configurational entropy of the alloy system was estimated from the elemental composition obtained through XPS and ICPMS and the value exceeded the recommended HEA limiting entropy value of 1.54R (ESI†).
To understand the chemical environment of the metals in the HEACB heterostructure, a detailed high-resolution XPS spectra analysis of each specific element along with a wide scan was conducted, as shown in Fig. 2n–s and S7.† Charge correction was applied by calibrating the peaks with respect to the C 1s peak at 284.7 eV. The constituent metals were found to exist in a multi-valence state, as shown in Fig. 2. To compare the effect of heterojunction formation on the electronic structure of the HEA alloys in HEACB, we performed a comparative XPS analysis with that of bare HEA as shown in Fig. S8.† One could observe a slight shift in the binding energy towards a higher value in the case of HEACB with respect to the bare HEA. This indicates synergistic electron diffusion at the alloy–carbon interface in HEACB. Electrons are induced from the HEA towards the carbon support, resulting in a binding energy shift of the constituent metals. There have been reports on similar interactions in such heterostructures, where the 3d orbitals can effectively interact with 2p orbitals of carbon atoms on the supporting matrix, thereby tuning their electronic band structure, providing a continuous adsorption energy for various intermediates for efficient catalysis.47,48,51,52 The effect of interaction of HEA nanoparticles with carbon is also reflected in the thermogravimetric analysis (TGA). The TGA study in an oxygen atmosphere (Fig. S9†) shows a gradual mass loss for HECB 1000 (∼600 to 700 °C) in contrast to the carbon black. In the case of bare carbon black, we could see a sharp degradation around 500 °C. The gradual decrease is due to the interaction of HEAs with the carbon support. The presence of oxygen was observed in the HEACB particles as seen in the elemental mapping shown in Fig. S10.† The absence of metallic states of Ni, Cr, and Fe could be due to the surface oxidation happening during sample handling, as well as the strong interaction of HEA with CB or due to the presence of adsorbed oxygen on the catalyst surface. The resolved O 1s spectrum (Fig. S11†) and the Raman spectrum scanned in the lower wavenumber range (Fig. S12†), reveal the absence of lattice oxygen and M–O bonds, with the major oxygen contribution originating from the surface adsorbed moisture and similar components.53,54
Furthermore, XPS was employed to calculate the surface elemental composition based on HR XPS (Table S4†) to confirm the HEA nature of the surface and the corresponding configurational entropy factor was calculated to be 1.54R which falls within the classified range for HEAs.41 To conclude the material characterization of HEACB, the single-FCC crystal phase formation, as correlated with XPS, ICPMS, and STEM-EDS analysis, confirms that the HEA quintuple alloying was successful on the carbon support.
3.2. Sodium borohydride hydrolysis
The catalytic activity of the composite alloy was evaluated by dispersing 50 mg of the catalyst in 100 mL of a 0.1 M NaBH4 solution and by measuring the amount of hydrogen produced during hydrolysis at room temperature. Fig. 3a shows the rate of hydrogen evolution during the NaBH4 hydrolysis in the presence of HEACB 500, HEACB 750, and HEACB 1000, and a comparison with CB and bare HEA. It may be noted that the time taken to generate 100 mL of hydrogen gas is ∼690, 870, 1088, 1411 and 2244 s for HEACB 1000, HEACB 750, HEACB 500, bare HEA and only carbon black, respectively. Similarly, the TOF is 188, 146, 112, and 43 mL H2 gMn−1 s−1 for HEACB 1000, HEACB 750, HEACB 500, and bare HEA, respectively (Fig. 3b) suggesting the superior catalytic activity of HEACB 1000 over other catalysts. This indicates that HEACB could catalyze the reaction more efficiently than bare HEA. This could be due to the chemical environment modulation of the HEA nanoparticles by the supporting carbon matrix owing to the interaction of vacant d orbitals of the 3d metals in HEA NPs with the 2p orbital of carbon, thus altering the mode of adsorption of intermediates onto HEACB and hence the effective work function of the heterostructure, which will be discussed in detail in the computational section. To validate the direct involvement of the metal centers of HEA in the active catalyst cycle, we performed a thorough poison test for all the catalysts, keeping all other experimental parameters constant. The effect of the poison on hydrogen production was studied at the catalysts by adding 10 mL of 0.1 M NaCN to the prepared NaBH4 solution. Fig. 3c and S13† show the effect of the poison on the NaBH4 hydrolysis rate at HEACB 500, HEACB 750, and HEACB 1000 catalysts (about 50 mg) with and without adding NaCN to the reaction medium at 60 °C. For the production of 100 mL of H2, it takes 325, 265, and 225 s for HEACB 500, HEACB 750, and HEACB 1000 before NaCN addition. After adding NaCN, it takes 585, 587, and 595 s to produce the same amount of hydrogen at HEACB 500, HEACB 750, and HEACB 1000 catalysts, respectively. This rapid reduction in activity is due to the blockage of active metal centers by potent cyanide ligands, which is reflected in the values of TOF, as represented in the bar plot in Fig. 3d. This indicates the direct involvement of HEA active metal centers in catalysis. | ||
| Fig. 3 NaBH4 hydrolysis and Mott–Schottky analysis: (a) rate comparison of hydrogen evolution during NaBH4 hydrolysis using different HEACB catalysts, (b) TOF of HEACBs towards NaBH4 hydrolysis represented as a bar plot, (c) cyanide poisoning test on the HEACB CB 1000 sample, (d) bar plot showing the impact of cyanide poisoning on the TOF values of HEACB samples, (e) temperature dependent rate study of HEACB 1000 for NaBH4 hydrolysis, (f) Arrhenius plot of HEACB 1000, HEACB 750, and HEA 500, (g) mass loading study conducted for HEACB 1000 towards the hydrolysis and (h) band structure of HEACB at the heterostructure–electrolyte interface under flat band conditions and (i) Fermi level equilibrated conditions. | ||
To gain further insight into the reason behind the difference in the reaction rates and feasibility of hydrogen production, temperature-dependent studies have been carried out in the presence of 100 mg of catalysts and 0.1 M NaBH4. Fig. 3e and S14† show the effect of temperature on the hydrogen production rate, which implicitly reflects the net activation barrier of the reaction. In all the cases, the hydrogen production rates increase with the temperature, which verifies the Arrhenius behavior. Fig. 3f and S15† show the Arrhenius plots of NaBH4 hydrolysis with HEACB catalysts. The activation energy for bare HEA, HEACB 500, HEACB 750, and HEACB 1000 is found to be 43, 41, 22, and 15 kJ mol−1, respectively. The lower activation energy of HEACB 1000 suggests that this heterostructure is more effective in reducing the energy barrier of NaBH4 hydrolysis indicating a better redox process and electron transfer by offering better and optimal intermediate adsorption and charge transfer, which has been discussed in detail in the DFT section.55
Fig. 3g shows the effect of catalyst loading with 25, 50, and 100 mg of material on the reaction rate at room temperature. We could observe a direct proportionality between activity and catalyst mass loading. Fig. S15c† shows the plot of time required for the production of 100 mL of H2vs. catalyst loading. The higher the loading, the more catalytic active centers of HEAs are available to enhance the hydrolysis process and reduce the time required to produce the required amount of hydrogen, which holds more practical significance. This indicates the high hydrogen turnover from HEACB 1000 and its least incubation time needed for the reaction to start compared to HEACB 750 and 500. This can be directly correlated to the degree of exposed active metal centers and the total surface area of the heterostructure.46,56 Faster adsorption of the intermediates and the reactant molecules favored by the high degree of exposed active centers results in a fast response of the catalyst towards active hydrogen production.57
3.3. Mott–Schottky analysis
We employed electrochemical Mott–Schottky (M–S) analysis to gain deeper insights into the band structure at the interface of these materials.26 Since the HEA nanoparticles are dispersed onto a conductive carbon support, we anticipate semiconductor behavior at the reaction interface.26,44,48 The formation of this interface is expected to induce electron flow from the electron-rich metal-semiconductor system to the less electron-rich electrolyte until the Fermi levels reach equilibrium, as illustrated in Fig. 3h and i. To confirm this phenomenon, we conducted a detailed M–S analysis (Fig. 4a and b) at a constant current frequency of 1 kHz and an amplitude of 20 mV, within a potential range of −0.2 to 1.1 V vs. RHE. The linear relation observed for the inverse capacitance squared versus potential difference signifies the presence of a semiconductor–electrolyte junction in our system, further evidenced by the presence of three distinct potential regions as shown in Fig. 4a. The positive slope in the graph suggests that our catalysts behave as n-type semiconductors compared to the electrolyte. A key parameter we can extract is the flat band potential (Vfb), represented by the x-axis intercept in Fig. 4b.58 If we compare the three HEACB samples, we can clearly see a considerable shift in the flat band potential towards the positive side of potential for HEACB 1000 compared to HEACB 750 and HEACB 500. This positive shift indicates that the Mott–Schottky junction formed in the case of HEACB 1000 has reduced band bending, thus favoring faster charge transfer kinetics across the interface. The analysis also revealed that HEACB 1000 exhibits an enhanced charge carrier density compared to the other two samples. It is observed that on integrating conductive carbon into the HEA nano alloys, the carrier density enhanced which could be expected due to the increased electron availability because of the conductive support at the reaction interface; this enhanced electron availability improves the probability of intermediate adsorption and facilitates faster electron transfer kinetics.59–61 When the MS plot was compared between HEACB and bare HEA, a significant shift in the flat band potential towards the positive direction was observed when the heterostructure was formed in comparison to the bare HEA nanoparticles. This can be explained based on the tendency of electronic flow from HEA particles towards the carbon support at their heterojunction interface, which lowers the Fermi level of the system and thus reduces the band bending of the catalyst with respect to the electrolyte system, hence favoring better kinetics during catalysis. This observation could explain the reason for the enhanced catalytic efficiency improvement observed upon carbon–HEA heterostructure formation. The XPS analysis of HEACB and bare HEA also supports this finding, where in the case of HEA bare, the metal peaks are slightly shifted to the lower binding energy compared to HEACB, indicating a synergistic interaction existing between HEA and CB. | ||
| Fig. 4 Electrochemical analysis of HEACB samples: (a) MS plot of the HEACB 1000 heterostructure in 0.5 M KOH medium electrolyte indicating regions of band interface modulation, (b) MS plot comparison of HEACB catalysts, (c) LSV of HEACB samples in the NaBH4 oxidative polarization region in 0.3 M NaBH4 and 0.5 M KOH electrolyte, (d) Tafel slope analysis, (e) NaBH4 concentration optimization study, (f) diffusion study, (g) Randles–Sevcik plot of current vs. root of scan rate, (h) electrochemical impedance spectroscopic analysis of HEACB in 0.5 M KOH electrolyte (inset: equivalent circuit), and (i) radar plot showing the comparison of various catalytic parameters. | ||
3.4. Electrochemical analysis of NaBH4-mediated H2 evolution
During both the chemical and electrochemical evolution of hydrogen from NaBH4, the NaBH4 ultimately gets oxidised. Therefore, we have attempted to probe the kinetics of the oxidation process of NaBH4 in a water reaction medium through electrochemical investigation. The electrochemical activity of all the HEACB catalysts was evaluated through linear sweep voltammetry (LSV) in 0.3 M NaBH4 solution and 1 M NaOH as electrolyte at a scan rate of 10 mV s−1 as shown in Fig. 4c. As a control experiment, we first studied bare GC without any catalyst coating, which showed no activity toward NaBH4 oxidation due to the absence of catalytic sites. When we modified the GC with the HEACB catalyst, we could see an increase in the current in the oxidative polarization region of −0.65 V to 0 V vs. Ag/AgCl. Concerning the standard oxidation potential of NaBH4 of about −1.03 V, we have estimated the overpotential of each HEACB catalyst. HEACB 1000 showed the lowest overpotential of 0.51 V, followed by HEACB 750 and HEACB 500 with values of 0.59 and 0.62 V, respectively, to drive a current density of 10 mA cm−2. The bare HEA showed the least activity, as observed in the NaBH4 hydrolysis part, suggesting the importance of the carbon support in enhancing the overall catalytic activity. This indicates the efficacy of HEACB 1000 in favoring NaBH4 oxidation by reducing the effective energy barrier, which was also reflected in the hydrolysis experiment of NaBH4. To gain deeper insight into the kinetics of the reaction, we performed a Tafel slope analysis on the kinetic region of LSV near the onset potential of HEACB catalysts as shown in Fig. 4d. The Tafel slope is inversely proportional to the charge transfer coefficient of a particular reaction as per the Butler–Volmer equation. Hence, a lower slope signifies a faster charge transfer.62 The magnitude of the Tafel slope follows the order of HEACB 1000 < HEACB 750 < HEACB 500 < bare HEA, which is consistent with the trend we experimentally observed during the kinetic study of chemical hydrogen evolution via NaBH4 oxidation. This indicates that a common active site is responsible for the oxidation of NaBH4 in both chemical and electrochemical hydrogen evolution pathways. Moreover, when we correlated the activation energy estimated by the temperature dependent hydrolysis studies with the electrochemically extracted Tafel slope (Fig. S15d†), we could see a linear correlation. Similarly, we could see an exponential relation between the rate of reaction (TOF) and the Tafel slope (Fig. S15e†), which is in accordance with the Butler–Volmer equation,62 which also supports the inherent relation existing between the chemical and electrochemical hydrolysis of the NaBH4 process. The electrochemical active surface area (ECSA) comparison of HEACB and bare HEA as shown in Fig. S16 and Table S5,† also suggests that the carbon black–HEA heterostructure formation helps in exposing potential sites and thereby enhancing the efficiency of electrocatalysis.The electrochemical oxidation of NaBH4 is a diffusion-limited process. The current rate is expected to depend primarily on the concentration of oxidative species NaBH4 since the diffusion rate is dependent on the effective concentration gradient of the reactant on the electrode surface. Fig. 4e shows the NaBH4 concentration optimization analysis, where we can see an initial increase in the reaction rate with NaBH4 concentration from 0.1 to 0.4 M after which it reaches saturation. This is where diffusion limitations can no longer be suppressed by simple concentration variation within the studied potential region. To verify the diffusion-limited process further, we have experimentally reproduced the Randles–Sevcik equation.63 For that, a similar LSV experiment was conducted with varying scan rates, keeping all other experimental parameters unchanged, as shown in Fig. 4f. We could see a gradual increase in current, indicating the enhanced diffusion flux at higher scan rates on the electrode surface. The current density versus the square root of the scan rate, as shown in Fig. 4g, and the linear relationship clearly indicate that a diffusion-limited electro-oxidation process of NaBH4 occurs on the surface of HEACB catalysts.
Another important kinetic aspect of probing is the resistance of electron flow in the charge transfer regime of the reaction. To estimate the amount of charge transfer resistance experienced during the electro-oxidation of NaBH4, electrochemical impedance spectroscopic analysis (EIS) was employed as shown in Fig. 4h (inset shows the equivalent circuit of the system). The resistive components used in the equivalent circuit, R1, R2, and R3, indicate solution resistance, charge transfer resistance and adsorption resistance due to porosity of the material, respectively. The components Q1 and Q2 represent two constant phase elements due to the double layer formation and material porosity effects, respectively. The details and fitted values are listed in Table S6.† The diameter of the semicircular region in the plot corresponds to the charge transfer resistance.64,65 We could observe that the charge transfer resistance is minimum for the HEACB 1000 heterostructure and is maximum for bare HEA as expected. This could be due to the higher degree of FCC-phased HEA formation in the case of HEACB 1000. To summarize the entire analysis, we gained further insight into the reasons behind the experimental observations of NaBH4 hydrolysis through the estimation of the material's band structure at the reaction interface and by examining significant kinetic aspects like overpotential Tafel slope, donor density and charge transfer resistance. Fig. 4i shows the overall comparison of various reactions and catalytic parameters of all three HEACB heterostructures as a radar plot. This plot suggests that catalytic inherent parameters like impedance, Tafel slope, and flat band potential have a direct correlation with activity descriptors like turn over frequency, current density, activation energy and so on. We can gain further insight into these activity descriptors and the reasons behind their activity trend by analyzing the d band structure and interaction of the reaction intermediate on the heterostructure through theoretical calculations.
3.5. DFT calculation of NaBH4 hydrolysis on the HEA surface
We used density functional theory (DFT) simulations to understand why the composite HEA outperformed bare HEA in borohydride hydrolysis and the underlying cause behind the enhanced activity of the heterostructure. A defected graphene sheet was formed by removing two adjacent C-atoms from a 7 × 7 graphene supercell to model the amorphous carbon system. An icosahedral nanoparticle with 13 atoms and a random arrangement of five metals was tailored to simulate HEA nanoparticles (Fig. S17†). Each metal's composition was estimated using the experimental composition ratio (Table S2†). Carbon atoms near defective positions have longer C–C bonds than in pristine sheets. This causes instabilities in the carbon atoms, which bind to the sheet more loosely than a pure sheet. To achieve maximum stability, the created HEA nanoparticle was placed on top of the defective region of the graphene sheet, where the less tightly linked carbon can interact with the HEA metals (Fig. S17†). To reduce periodic image interactions, a 15 Å vacuum was applied perpendicularly. The composite catalyst system has a formation energy of −0.07 eV per atom, making it thermodynamically stable.There are numerous unique active sites on a HEA nanoparticle surface, making it challenging and time-consuming to identify the most active sites. To address this, we employed the d-band model, which successfully explains the trends in adsorption energy across multiple reactions.66 According to this approach, metals with low d-band centre values relative to the Fermi level are the most active sites. This results in fewer electrons in antibonding states and more electrons in bonding states created by the metal–adsorbate interaction. The average d-band center of the various metals found on the HEA surface for both composite and bare HEA nanoparticles are presented (Fig. S18†). According to the d-band center, the most active site in the bare HEA system is the Cr atom (Cr1), with the Mn atom (Mn3) coming in second. However, in the composite HEA, the tendency is reversed, the Mn atom (Mn4) is the most active site, while the Cr atom (Cr1) is the second most active. This observation demonstrates a significant interaction between the defective graphene and the HEA nanoparticles, which alters the electronic structure of the composite HEA compared to the bare HEA. To demonstrate the practicability of the aforementioned d-band method, the bare HEA system was considered. For NaBH4 hydrolysis, the BH4 unit must be adsorbed over the active site; thus, we evaluated the adsorption energy of boron-containing intermediates (BH4, BH3, and BH3OH) over the Cr and Mn active sites separately to compare the adsorption capabilities of the two active sites. Fig. S19† demonstrates the strong adsorption of distinct intermediates over the Cr active site rather than the Mn site, which is consistent with our d-band center approach. Fig. S19(b–g†) show the optimized structures of BH4, BH3, and BH3OH on the Cr and Mn sites, respectively.
Next, the adsorption of BH4− and OH2 over the active sites of both of the catalysts was studied. The adsorption energy of *BH4 + *OH2 on the heterostructure HEA and bare HEA are −3.26 eV and −2.97 eV, respectively (Fig. 5a). As a result, *BH4 + *OH2 is readily adsorbed and hence likely to be easily hydrolyzed on the composite HEA surface. To further confirm this, the kinetic barrier of the B–H bond dissociation has been calculated, which is one of the crucial steps.67 The composite HEA shows a low barrier (0.37 eV) for the B–H dissociation compared to bare HEA (1.15 eV). The low barrier of B–H dissociation on composite HEA is due to the stronger adsorption of the dissociated H on the three-fold hollow site (Fig. 5d). On the other hand, the dissociated H is adsorbed on the bridge site on the bare HEA surface (Fig. 5b). The partial density of states (PDOS) plots reveal the hybridization of the H 1s states with the 3d states of the corresponding adsorbent metal atoms (Fig. 5c–e). Subsequently, the adsorbed OH2 molecule can react with BH4−, releasing H2 molecules and forming BH3OH−.68–71
 | ||
| Fig. 5 (a) Adsorption energy of the intermediates. The optimized structure of *BH3 + *H + *OH2 over (b) bare HEA and (d) composite HEA. The adsorption of dissociated H is shown in a white circle for both the cases. PDOS plots for the dissociated H adsorption over the (c) bridge site in bare HEA and (e) three-fold hollow site in composite HEA. | ||
The adsorption energies of all the intermediates (*BH3 + *H + *OH2, *BH3 + *H + *OH + *H, and *BH3OH) up to the first H2(g) generation have been evaluated (Fig. 5a).70 The composite HEA always favors the adsorption of all the intermediates over the bare HEA. In the subsequent steps of the reaction, the borohydride's remaining H atoms are replaced by OH− ions, finally leading to the formation of B(OH)4− species.68,71 Subsequently, the computational findings support the experimental outcome where composite HEA outperformed bare HEA nanoparticles in borohydride hydrolysis. Thus, the combined experimental and computational studies highlight that the NaBH4 hydrolysis on the HEA heterostructure provides new scientific insights into blue hydrogen production. This work clearly demonstrates the promising potential of high entropy alloys as a commercially viable platform for sustainable hydrogen production and their effective utilization in the future.
4. Conclusions
The HEA-decorated conductive carbon heterostructure was successfully fabricated, and we found that higher temperatures favor the FCC-phased HEA stabilization. The HEACB 1000 catalyst showed a promising catalytic response towards the hydrolysis of NaBH4 for hydrogen fuel production. The catalyst was benchmarked with a TOF value of approximately 188 mL gMn−1 s−1 and a lesser activation energy of about 15 kJ mol−1. Mott–Schottky analysis revealed that the HEACB heterostructure exhibited n-type semiconductor behavior at the electrolyte interface during the reaction. The analysis indicated an optimal flat band potential and carrier density for the sample with a high concentration of HEA alloys. It was observed that on integrating conductive carbon into the HEA nanoalloys, the carrier density increased which could be expected due to the presence of electrons in the CB available at the reaction interface. This enhanced electron availability attenuates the probability of intermediate adsorption and facilitates faster electron transfer kinetics as observed in EIS analysis. Additionally, the electrochemical analysis provided more kinetic information about the NaBH4 oxidation, for instance, HEACB 1000 showed an overpotential of 0.51 V, a Tafel slope of 286 mV dec−1, and a charge transfer resistance of about 24 Ω cm−2, validating the catalytic efficiency of HEACB towards NaBH4 oxidation. DFT supported the experimental findings and revealed the potential active sites and adsorption modes of intermediates, justifying the enhanced activity of the HEACB composite over bare HEA NPs. This work underscores the potential of the HEACB heterostructure in achieving efficient catalytic activity for both chemical and electrochemical production of hydrogen fuel via NaBH4 hydrolysis towards a greener and more sustainable future.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
G. R.: conducted the majority of experiments and materials synthesis and analyzed structural and electrochemical data, manuscript writing, and editing. R. N.: material synthesis, conceptualization, editing of the manuscript. S. M. and A. K. S.: conducted DFT calculations, analyzed the results and contributed to manuscript writing and editing. K. K.: involved in synthesizing and characterizing samples and manuscript writing. K. K. N.: conceptualized the study, acquired project funding, supervision, scientific discussions, and project administration, and contributed to reviewing and editing the manuscript. All authors have collectively agreed upon the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. R. would like to thank the Ministry of Human Resource Development (MHRD) for the research scholarship. The authors also acknowledge the Department of Science and Technology (DST), India, for funding the BET adsorption–desorption facility (DST-ERB, EMR/2016/005843 and DST-FIST, SR/FST/PS11-9/2010) for the research. The authors also thank the Council of Scientific & Industrial Research (CSIR) (SP/CSIR-21-0002) for the project funding. G. R. expresses sincere gratitude to Dr Demudu Babu Gorle in the Materials Research Centre (MRC) for the insightful scientific discussion regarding electrochemical borohydride hydrolysis. G. R. thanks the Materials Research Centre for providing access to the XRD facility, the Centre for Nanoscience and Engineering (CeNSE), and the Inorganic and Physical Chemistry (IPC) department at IISc for granting access to the SEM and TEM facilities. S. M. expresses gratitude for the support received from the Prime Minister's Research Fellowship. Both S. M. and A. K. S. extend their thanks to the Materials Research Centre (MRC), Supercomputer Education and Research Centre (SERC), and Solid State and Structural Chemistry Unit (SSCU) at the Indian Institute of Science, Bangalore, for providing essential computational facilities. S. M. and A. K. S. also acknowledge the support from the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India (File Number: CRG/2021/000633).References
- K. de Kleijne, M. A. J. Huijbregts, F. Knobloch, R. van Zelm, J. P. Hilbers, H. de Coninck and S. V. Hanssen, Nat. Energy, 2024, 9, 1139–1152 CrossRef CAS
.
- Ş. Neaţu, F. Neaţu, I. M. Chirica, I. Borbáth, E. Tálas, A. Tompos, S. Somacescu, P. Osiceanu, M. A. Folgado, A. M. Chaparro and M. Florea, J. Mater. Chem. A, 2021, 9, 17065–17128 RSC
- J. Guo, C. K. Yin, D. L. Zhong, Y. L. Wang, T. Qi, G. H. Liu, L. T. Shen, Q. S. Zhou, Z. H. Peng, H. Yao and X. B. Li, ChemSusChem, 2021, 14, 2655–2681 CrossRef CAS PubMed
- H. N. Abdelhamid, Int. J. Hydrogen Energy, 2021, 46, 726–765 CrossRef CAS
- C.-C. Su, M.-C. Lu, S.-L. Wang and Y.-H. Huang, RSC Adv., 2012, 2, 2073–2079 RSC
- C. Wang and D. Astruc, Chem. Soc. Rev., 2021, 50, 3437–3484 RSC
- M. Dragan, Catalysts, 2022, 12, 356 CrossRef CAS
- F. Xu, J. Ren, J. Ma, Y. Wang, K. Zhang, Z. Cao, Q. Sun, S. Wu, G. Li and S. Bai, Int. J. Hydrogen Energy, 2024, 50, 827–844 CrossRef CAS
- A. M. Ozerova, A. A. Skobelkina, V. I. Simagina, O. V. Komova, I. P. Prosvirin, O. A. Bulavchenko, I. L. Lipatnikova and O. V. Netskina, Materials, 2022, 15, 3010 CrossRef CAS PubMed
- J. Chen, Z.-H. Lu, W. Huang, Z. Kang and X. Chen, J. Alloys Compd., 2017, 695, 3036–3043 CrossRef CAS
- N. Sahiner, O. Ozay, E. Inger and N. Aktas, J. Power Sources, 2011, 196, 10105–10111 CrossRef CAS
- H. Wang, F. Xu, L. Sun, J. Wu, G. Zhang, Y. Zhu, Q. Shao, Y. Luo, X. Peng, Y. Wang, Y. Gao and Y. Zou, J. Alloys Compd., 2024, 978, 173415 CrossRef CAS
- T. Li, C. Xiang, Y. Zou, F. Xu and L. Sun, J. Alloys Compd., 2021, 885, 160991 CrossRef CAS
- D. D. Megersa, Y. Kim, N. Kim, J. Lee, J. S. Bae, J.-Y. Choi and H. K. Yu, Int. J. Hydrogen Energy, 2022, 47, 15687–15694 CrossRef CAS
- S. Fang, Y. Chen, S. Wang, J. Xu, Y. Xia, F. Yang, Y. Wang, J. Lao, C. Xiang, F. Xu, L. Sun, Y. Zou and H. Pan, J. Alloys Compd., 2023, 936, 167990 CrossRef CAS
- W. Zhang, W. Yuan, X. Zhang, Y. Ke, Y. Wu, Y. Bai, S. Jiang and Y. Tang, J. Mater. Chem. A, 2024, 12, 18705–18732 RSC
- C. Gao, S. Wang, X. Liu and C. V. Singh, Mater. Adv., 2024, 5, 4231–4241 RSC
- F. Liu, P. K. Liaw and Y. Zhang, Metals, 2022, 12, 501 CrossRef CAS
- L. Kong, B. Cheng, D. Wan and Y. Xue, Front. Mater., 2023, 10 DOI:10.3389/fmats.2023.1135864
- R. K. Pittkowski, C. M. Clausen, Q. Chen, D. Stoian, W. van Beek, J. Bucher, R. L. Welten, N. Schlegel, J. K. Mathiesen, T. M. Nielsen, J. Du, A. W. Rosenkranz, E. D. Bøjesen, J. Rossmeisl, K. M. Ø. Jensen and M. Arenz, EES Catal., 2023, 1, 950–960 RSC
- S. Kord, F. Fathirad, D. Afzali and M. Fayazi, Inorg. Chem. Commun., 2022, 136, 109130 CrossRef CAS
- S. Duman and S. Özkar, Int. J. Hydrogen Energy, 2018, 43, 15262–15274 CrossRef CAS
- Y. Wei, M. Wang, W. Fu, S. Si, L. Wei, X. Zhao and Y. Wang, Mater. Lett., 2022, 308, 131166 CrossRef CAS
- Y. Xia, Y. Pei, Y. Wang, F. Li and Q. Li, Fuel, 2023, 331, 125733 CrossRef CAS
- J. Hao, Z. Zhuang, K. Cao, G. Gao, C. Wang, F. Lai, S. Lu, P. Ma, W. Dong, T. Liu, M. Du and H. Zhu, Nat. Commun., 2022, 13, 2662 CrossRef CAS PubMed
- G. Raj, R. Nandan, K. Kumar, D. B. Gorle, A. B. Mallya, S. M. Osman, J. Na, Y. Yamauchi and K. K. Nanda, Mater. Horiz., 2023, 5032–5044 RSC
- M. Bondesgaard, N. L. N. Broge, A. Mamakhel, M. Bremholm and B. B. Iversen, Adv. Funct. Mater., 2019, 29, 1905933 CrossRef CAS
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS
- D. Gazzoli, M. Occhiuzzi, A. Cimino, G. Minelli and M. Valigi, Surf. Interface Anal., 1992, 18, 315–322 CrossRef CAS
- J. Jin, R. Gao, H. Peng, H. Guo, S. Gong and B. Chen, Metall. Mater. Trans. A, 2020, 51, 2411–2429 CrossRef CAS
- Y. Zhang, Y. Zhu, Y. Cao, D. Li, Z. Zhang, K. Wang, F. Ding, X. Wang, D. Meng, L. Fan and J. Wu, RSC Adv., 2016, 6, 81989–81994 RSC
- M. Aizenshtein and S. Hayun, Metallogr., Microstruct., Anal., 2020, 9, 305–311 CrossRef CAS
- Y. Ma, B. Jiang, C. Li, Q. Wang, C. Dong, P. K. Liaw, F. Xu and L. Sun, Metals, 2017, 7, 57 CrossRef
- S. Banerjee, A. Kumar, P. Ruz and P. Sengupta, Int. J. Hydrogen Energy, 2016, 41, 18130–18140 CrossRef CAS
- W. Wang, W. Qi, L. Xie, X. Yang, J. Li and Y. Zhang, Materials, 2019, 12, 694 CrossRef CAS PubMed
- B. L. Checa Fernández, J. M. Martín, G. Sarriegui and N. Burgos, J. Mater. Res. Technol., 2023, 24, 1454–1467 CrossRef
- S. Kumar, D. Tripathi, A. Anshu, R. Kumar, A. Yadav, R. K. Rawat, P. Chauhan and B. Sahoo, ACS Appl. Nano Mater., 2024, 7, 21603–21616 CrossRef CAS
- G. Raj, R. Nandan, P. Gakhad, K. Kumar, A. K. Singh and K. K. Nanda, Chem. Eng. J., 2024, 158041 Search PubMed
- O. Y. Bisen, R. Nandan, G. Raj, A. K. Yadav and K. K. Nanda, ACS Appl. Energy Mater., 2022, 5, 14019–14034 CrossRef CAS
- R. Nandan, G. Raj and K. K. Nanda, ACS Appl. Mater. Interfaces, 2022, 14, 16108–16116 CrossRef CAS PubMed
- G. Raj, D. Das, B. Sarkar, S. Biswas and K. K. Nanda, Sustainable Mater. Technol., 2022, 33, e00451 CrossRef CAS
- B. Roul, D. B. Gorle, G. Raj, K. Kumar, M. Kumari, K. K. Nanda and S. B. Krupanidhi, J. Mater. Chem. C, 2023, 11072–11081 RSC
- K. Kumar, H. Rachna Devi, G. Raj and K. Kar Nanda, J. Mater. Chem. C, 2024, 12, 4026–4036 RSC
- K. Kumar, P. Tripathi, G. Raj, D. Kalyan, D. B. Gorle, N. G. Mohan, S. K. Makineni, K. Ramanujam, A. K. Singh and K. K. Nanda, Green Chem., 2024, 7931–7943 RSC
-
S. S. Naboychenko, I. B. Murashova and O. D. Neikov, Handbook of Non-Ferrous Metal Powders, ed. O. D. Neikov, S. S. Naboychenko, I. V. Murashova, V. G. Gopienko and I. V. Frishberg, Elsevier, Oxford, 2009, pp. 436–484 Search PubMed
- S. Li, D. Leng, W. Li, L. Qie, Z. Dong, Z. Cheng and Z. Fan, Energy Storage Mater., 2020, 27, 279–296 CrossRef
- K. Zeng, J. Zhang, W. Gao, L. Wu, H. Liu, J. Gao, Z. Li, J. Zhou, T. Li, Z. Liang, B. Xu and Y. Yao, Adv. Funct. Mater., 2022, 32, 2204643 CrossRef CAS
- R. Nandan, M. Y. Rekha, H. R. Devi, C. Srivastava and K. K. Nanda, Chem. Commun., 2021, 57, 611–614 RSC
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. De Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS
- J. Zhang, H. Liu, Y. Gu, J. Zhang, X. Zhang and X. Qi, J. Mater. Sci.:Mater. Electron., 2022, 33, 9918–9929 CrossRef CAS
- B. Wang, Y. Yao, X. Yu, C. Wang, C. Wu and Z. Zou, J. Mater. Chem. A, 2021, 9, 19410–19438 RSC
- L. Zhang, Q. Luo, X. Chen, M. S. Tse, O. K. Tan, K. H. H. Li, Y. Y. Tay, C. K. Lim, X. Guo and H. holden, RSC Adv., 2016, 6, 65038–65046 RSC
- W. Luo, Y. Wang, L. Luo, S. Gong, Y. Li and X. Gan, Appl. Surf. Sci., 2022, 606, 154808 CrossRef CAS
- H. R. Devi, B. C. Ong, X. Zhao, Z. Dong, K. K. Nanda and Z. Chen, Energy Technol., 2022, 10, 2100457 CrossRef CAS
- H. Zhong, C. Yang, L. Fan, Z. Fu, X. Yang, X. Wang and R. Wang, Energy Environ. Sci., 2019, 12, 418–426 RSC
- B. Sarkar, D. Das and K. K. Nanda, J. Mater. Chem. A, 2021, 9, 13958–13966 RSC
- J. Hou, Y. Sun, Y. Wu, S. Cao and L. Sun, Adv. Funct. Mater., 2018, 28, 1704447 CrossRef
- T. T. Yang and W. A. Saidi, J. Phys. Chem. Lett., 2022, 13, 5310–5315 CrossRef CAS PubMed
- D. Saranya, D. Velayutham and V. Suryanarayanan, J. Electroanal. Chem., 2014, 734, 70–78 CrossRef CAS
- S. Kumar, R. Kumar, N. Goyal, A. Vazhayil, A. Yadav, N. Thomas and B. Sahoo, ACS Appl. Nano Mater., 2024, 7, 7865–7882 CrossRef CAS
- S. Kumar, R. Kumar, N. Goyal, A. Yadav, S. BM and B. Sahoo, ACS Appl. Nano Mater., 2024, 7, 16422–16437 CrossRef CAS
- W.-P. Zhou, X. Yang, M. B. Vukmirovic, B. E. Koel, J. Jiao, G. Peng, M. Mavrikakis and R. R. Adzic, J. Am. Chem. Soc., 2009, 131, 12755–12762 CrossRef CAS PubMed
- A. Leelavathi, R. Ahmad, A. K. Singh, G. Madras and N. Ravishankar, Chem. Commun., 2015, 51, 16856–16859 RSC
- B. Raju Karimadom, S. Varshney, T. Zidki, D. Meyerstein and H. Kornweitz, ChemPhysChem, 2022, 23, e202200386 CrossRef
- A. A. Kassem, H. N. Abdelhamid, D. M. Fouad and S. A. Ibrahim, Int. J. Hydrogen Energy, 2019, 44, 31230–31238 CrossRef CAS
- S. Zhou, L. Cheng, Y. Huang, Y. Liu, L. Shi, T. T. Isimjan and X. Yang, Appl. Catal., B, 2023, 328, 122519 CrossRef CAS
- A. E. Genç, A. Akça and B. Kutlu, Int. J. Hydrogen Energy, 2018, 43, 14347–14359 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06193d |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |