Ellagitannin biosynthesis: oxidation of pentagalloylglucose to tellimagrandin II by an enzyme from Tellima grandiflora leaves
Ruth
Niemetz
a,
Gerhard
Schilling
b and
Georg G.
Gross
*a
aMolekulare Botanik, Universität Ulm, 89069, Ulm, Germany.. E-mail: ruth.niemetz@biologie.uni-ulm.de;
georg.gross@biologie.uni-ulm.de
bUniversität Heidelberg, Organisch-Chemisches Institut, Im Neuenheimer Feld 270, D-69120, Heidelberg, Germany.. E-mail: gerhard.schilling@urz.uni-heidelberg.de
First published on 11th December 2000
Abstract
First evidence of the in vitro oxidation of 1,2,3,4,6-pentagalloylglucose to the ellagitannins, tellimagrandin II and 1,4,6-tri-O-galloyl-2,3-O-hexahydroxydiphenoyl-β- D-glucose, has been obtained with a partially purified enzyme from leaves of Tellima grandiflora (fringe cups, Saxifragaceae).
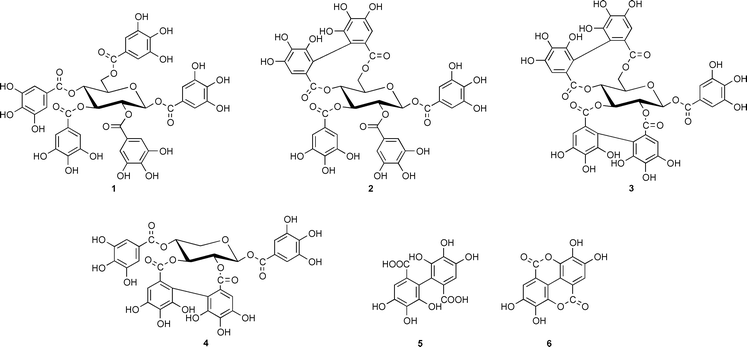
It was already recognized in the mid 1950’s that ellagitannins, a widespread subclass of hydrolyzable plant tannins, were characterized by 3,4,5,3′,4′,5′-hexahydroxydiphenic (HHDP) acid residues (5) that occur in Nature in the form of various glucose esters and which, after hydrolytic release, spontaneously rearranged to the dilactone, ellagic acid (6). It was discussed very early on that the biogenesis of such ellagitannins in plants should originate from 1,2,3,4,6-penta-O-galloyl-β-D-glucose (1) as the principal precursor,1 a view that was corroborated and refined later by Haslam and coworkers. It was proposed by these authors that the energetically preferred 4C1 conformer of 1 was sequentially oxidized to tellimagrandin II (2) and casuarictin (3).2,3 The recently recognized role of ellagitannins as promising chemotherapeutic agents4,5 has stimulated remarkable success in the challenge of chemically synthesizing such compounds.6,7 The biosynthesis of ellagitannins, in contrast, still remained completely obscure. The present situation was illustrated in a recent review article8 with the statement that ‘the in vitro biaryl coupling of 1 has yet to be achieved by an isolated enzyme.’ In several attempts to elucidate the mechanism of such a transition with the enzyme systems laccase/O2 or peroxidase/H2O29 only free ellagic acid (6) had been detected, while the formation of true ellagitannins, characterized by glucose-bound HHDP residues, has never been achieved.
We concluded that inadequate analytical techniques represented the decisive obstacle in such investigations, which suffered from minimal enzyme reaction rates yielding numerous structurally closely related reaction products and unspecific by-products, and particularly from the inherent risk of contamination with in vivo formed ellagitannins that had been transferred into the enzyme assays by complexation with proteins. Such problems are eliminated by using radioactively labeled compounds, a technique that dramatically increases both the sensitivity and specificity of test systems. We therefore produced [U-14C]pentagalloylglucose by photoassimilation of 14CO2 in leaves of the gallotannin synthesizing plant Rhus typhina (staghorn sumac) in >99% purity.10 This compound was used as standard substrate in an extended screening program for enzymes that formed reaction products liberating [14C]ellagic acid (6) upon hydrolysis, thus providing a general probe for the in vitro synthesis of ellagitannins of widely differing structures.
By this strategy we were able to discover a novel soluble enzyme in leaves of Tellima grandiflora (Pursh) Lindley (fringe cups, Saxifragaceae), a weed that is known as a rich source of ellagitannins. The partially purified protein11 was found to catalyze the conversion of [U-14C]pentagalloylglucose to several radioactively labeled products, while no reaction occurred in the presence of denatured enzyme (Fig. 1). The most prominent peak among these compounds coincided with authentic tellimagrandin II (2) in two different HPLC systems with acetonitrile and MeOH gradients, respectively. This fraction was isolated and hydrolyzed (4 M HCl, 100 °C, 4 h) to afford glucose, gallic acid and ellagic acid (6) as sole 14C-labeled degradation products as determined by HPLC and liquid-scintillation counting of the eluates, thus indicating the in vitro synthesis of a true ellagitannin.
![RP-18 HPLC analysis of in vitro formed oxidation products of
[14C]pentagalloylglucose with an enzyme from Tellima
grandiflora leaves. Assay mixtures, containing 12.5 μg (2,500 dpm)
1 and enzyme (4 pkat) in 50 μl HEPES buffer (50 mM, pH 5.0),
were incubated at 30 °C for 60 min, stopped by heat-denaturing of
enzyme, and analyzed by RP-18 HPLC. (—), Enzyme assay; (⋯),
blank with acid-denatured enzyme. (1), Pentagalloylglucose;
(2) tellimagrandin II. HPLC conditions: Reprosil NE, 5 μm, 250
× 4 mm i.d.; solvent A = 0.05% aq. H3PO4, B
=0.05% H3PO4, in MeOH; gradient 0–1 min 10% B,
1–3 min 10–30% B, 3–20 min 30–40% B, 20–40
min 40% B; flow rate 0.7 ml min−1. Radioactivity was
determined by fractionation of eluates and subsequent liquid-scintillation
counting.](/image/article/2001/CC/b006270g/b006270g-f1.gif) | ||
| Fig. 1 RP-18 HPLC analysis of in vitro formed oxidation products of [14C]pentagalloylglucose with an enzyme from Tellima grandiflora leaves. Assay mixtures, containing 12.5 μg (2,500 dpm) 1 and enzyme (4 pkat) in 50 μl HEPES buffer (50 mM, pH 5.0), were incubated at 30 °C for 60 min, stopped by heat-denaturing of enzyme, and analyzed by RP-18 HPLC. (—), Enzyme assay; (⋯), blank with acid-denatured enzyme. (1), Pentagalloylglucose; (2) tellimagrandin II. HPLC conditions: Reprosil NE, 5 μm, 250 × 4 mm i.d.; solvent A = 0.05% aq. H3PO4, B =0.05% H3PO4, in MeOH; gradient 0–1 min 10% B, 1–3 min 10–30% B, 3–20 min 30–40% B, 20–40 min 40% B; flow rate 0.7 ml min−1. Radioactivity was determined by fractionation of eluates and subsequent liquid-scintillation counting. | ||
Under the conditions given in Fig. 1, the enzyme reaction proceeded linearly for 30 min and had pH and temperature optima at pH 5 and 45 °C, respectively. Normal Michaelis–Menten kinetics were observed for the substrate, pentagalloylglucose (1), up to a maximal concentration of 320 μM, while increasing substrate inhibition occurred above this value. Replots of this data according to Lineweaver-Burk revealed a Km value of 110 μM (Vmax = 33 pkat [0.12 μmol h−1]).
For the unequivocal determination of the structure of the reaction product, 100 mg of unlabeled pentagalloylglucose (1), chemically synthesized from triacetylgalloylchloride and β-D-glucose12) was incubated in a scaled-up enzyme assay, affording 1.7 mg pure reaction product of >93% purity after semi-preparative HPLC.13 Negative FAB-MS of this substance revealed prominent peaks for the deprotonated molecular ion [M − H]− at m/z 937 (tellimagrandin II 2, Mr 938) and m/z 635 (trigalloylglucose, Mr 636).14 Proton NMR spectroscopy (500 MHz) displayed signals that corresponded to those of an authentic sample of tellimagrandin II (2). In particular, three characteristic singlets at δ ppm (TMS) 6.91 (2H), 6.94 (2H) and 7.05 (2H) were detected in d4-MeOH that corresponded to the aryl-2,6 hydrogens at C-1, C-2 and C-3 of 2, respectively, while the singlets at δ 6.47 (1H) and 6.60 (1H) were due to the 2,2′-hydrogens of diphenic acid (5) bound at C-4,6. Also the chemical shifts for the glucose moiety were in full agreement with those of authentic 2.
The reaction product displayed, however, strong additional singlets at 6.61 (1H) and 6.63 (1H) ppm that apparently were due to a different compound with a C-2,3 linked HHDP unit, as concluded by comparison with 1H NMR data from authentic samples of 2,3-O-hexahydroxydiphenoylglucose, casuarictin (3) and pedunculagin (structure as 3, but with a free anomeric OH-group at C-1). Evidently, the enzyme preparation had catalyzed the simultaneous synthesis of both tellimagrandin II (2) and isomeric 1,4,6-tri-O-galloyl-2,3-O-hexahydroxydiphenoyl-β- D-glucose (4) that had not been separated by RP-HPLC. This latter, unusual ellagitannin is not known as a natural product; it has been recently obtained, however, by total synthesis in two enantiomerically pure forms, mahtabin A and pterocarinin C, characterized by 2,3-R- and S-HHDP residues, respectively.15,16 (An earlier report proposing this structure for a compound named cercidinin A from Cercidiphyllum japonicum17 has been questioned by these authors.) Detection of such a compound raises questions about the specificity of the enzyme(s) catalyzing such transformations that prompt considerations on the eventual cooperation of product-specificity guiding enzymes, analogous to the recently reported ‘dirigent’ protein involved in lignan biosynthesis.18
In summary, first evidence has been provided by our results that the long sought clue to unravel the old enigma of ellagitannin biosynthesis is now available.
We thank Angelika Müller (Ulm) for excellent technical assistence, Dr G. Schmidtberg (Ulm) for MS analyses, and Dr A. Scalbert (Clermont-Ferrand, France) and Professor T. Yoshida (Okayama, Japan) for reference samples of ellagitannins. Financial support from the Deutsche Forschungsgemeinschaft (Bonn), the Fonds der Chemischen Industrie (Frankfurt/M) and from research grants of the University of Ulm is gratefully acknowledged.
Notes and references
- O. Th. Schmidt and W. Mayer, Angew. Chem., 1956, 68, 103 CAS.
- R. K. Gupta, S. M. K. Al-Shafi, K. Layden and E. Haslam, J. Chem. Soc., Perkin Trans. 1, 1982, 2525 RSC.
- E. A. Haddock, R. K. Gupta, S. M. K Al-Shafi, K. Layden, E. Haslam and D. Magnolato, Phytochemistry, 1982, 21, 1049 CrossRef CAS.
- K. S. Feldman, K. Sahasrabudhe, R. S. Smith and W. J. Scheuchenzuber, Bioorg. Med. Chem. Lett., 1999, 9, 985 CrossRef CAS.
- M. N. Clifford and A. Scalbert, J. Sci. Food Agric., 2000, 80, 1118 CrossRef CAS.
- K. S. Feldman, K. Sahasrabudhe, S. Quideau, K. L. Hunter and M. D. Lawlor, in Plant Polyphenols 2. Chemistry, Biology, Pharmacology, Ecology, G. G. Gross, R. W. Hemingway and T. Higuchi, eds., Kluwer Academic/Plenum Publishers, New York, 1999, p. 101. Search PubMed.
- K. S. Feldman and K. Sahasrabudhe, J. Org. Chem., 1999, 64, 209 CrossRef CAS.
- R. F. Helm, L. Zhentian, T. Ranatunga, J. Jervis and T. Elder, in Plant Polyphenols 2. Chemistry, Biology, Pharmacology, Ecology, G. G. Gross, R. W. Hemingway and T. Higuchi, eds., Kluwer Academic/Plenum Publishers, New York, 1999, p. 83. Search PubMed.
- G. G. Gross, in Comprehensive Natural Products Chemistry. Vol. 3. Carbohydrates and Their Derivatives Including Tannins, Cellulose, and Related Lignins, B. M. Pinto, ed., Elsevier, Amsterdam, 1999, p. 799. Search PubMed.
- H. Rausch and G. G. Gross, Z. Naturforsch. C: Biosci., 1996, 51, 473 Search PubMed.
- Fresh leaves (80 g) of Tellima grandiflora were homogenized in liquid N2, extracted with 250 ml Tris-HCl (1.5 M, pH 8.0)–Na borate (0.2 M, pH 7.5) (1∶1, by vol.) and centrifuged (30000 × g, 30 min). The supernatant was depleted of phenolics by stirring with Amberlite XAD (20 min), filtered and fractionated with (NH4)2SO4. The 30–80% pellet was resuspended in HEPES buffer (50 mM, pH 6.0), desalted, concentrated by ultrafiltration and chromatographed on a Sephacryl S-300 (Pharmacia Biotech) column (40 × 2.4 cm i.d.) in HEPES buffer..
- G. G. Gross, Z. Naturforsch. C: Biosci., 1983, 38, 519 Search PubMed.
- The reaction mixture (400 ml vol., pH 5, containing 100 mg 1 and 850 mg protein) was incubated for 60 min at 30 °C. After stopping the reaction by heat-denaturing the enzyme, the mixture was extracted with EtOAc. The contents of the dried organic phase were subjected to semi-preparative RP-18 HPLC on Kromasil (5μ, 250 × 20 mm i.d.; gradient: solvent A = 0.05% aq. H3PO4, B = acetonitrile; 0–1 min 5% B, 1–2 min 5–18% B, then isocratic at 18% B; flow rate 22 ml min−1). Relevant fractions were immediately neutralized, depleted of organic solvent in vacuo, extracted with EtOAc and rechromatographed twice by RP-18 HPLC on Reprosil NE (5 μm, 250 × 8 mm i.d.; solvents and gradient as in Fig. 1; flow rate 2.4 ml min−1). The purified product was neutralized, depleted of MeOH and applied to a column of Sephadex LH-20 (60 × 12 mm i.d., in water). After rinsing with water to remove H3PO4, the product was eluted with MeOH and lyophylized, affording 1.7 mg material of >93% purity as determined by analytical HPLC under the conditions given in Fig. 1..
- R. Isobe, T. Tanaka, G. Nonaka and I. Nishioka, Chem. Pharm. Bull. (Tokyo), 1989, 37, 1748 Search PubMed.
- K. Khanbabaee and K. Lötzerich, Liebigs Ann., 1997, 1571 Search PubMed.
- K. Khanbabaee and K. Lötzerich, J. Org. Chem., 1998, 73, 8723 CrossRef.
- G. Nonaka, M. Ishimatsu, M. Ageta and I. Nishioka, Chem. Pharm. Bull. (Tokyo), 1989, 37, 50 Search PubMed.
- L. B. Davin, H. B. Wang, A. L. Crowell, D. L. Bedgar, D. M. Martin, S. Sarkanen and N. G. Lewis, Science, 1997, 275, 362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |