Crystal structures of
(R,R)-{[Ph(Me)CH]2NLi·pmdeta} and
{[PhC(![[double bond, length half m-dash]](https://www.rsc.org/images/entities/h2_char_e006.gif) CH2)NH]Na·pmdeta}2:
alkali metal amides derived from
(R,R)-bis(α-methylbenzyl)amine†
CH2)NH]Na·pmdeta}2:
alkali metal amides derived from
(R,R)-bis(α-methylbenzyl)amine†
Philip C. Andrews*, Peter J. Duggan, Melissa Maguire and Peter J. Nichols
Department of Chemistry, Monash University, Clayton, Melbourne, Victoria 3800, Australia.. E-mail: p.andrews@sci.monash.edu.au
First published on 18th December 2000
Abstract
The reaction of BunM (M = Li, Na) with
[(R,R)]-bis(α-methylbenzyl)amine in the presence of pmdeta
results in the formation of the chiral lithium amide complex
(R,R)-{[Ph(Me)CH]2NLi·pmdeta} and,
remarkably, the sodium enamide,
{[PhC(![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH2)NH]Na·pmdeta}2;
both compounds have been authenticated by single crystal X-ray
diffraction.
CH2)NH]Na·pmdeta}2;
both compounds have been authenticated by single crystal X-ray
diffraction.
Our knowledge of the structural chemistry of alkali metal amides is now firmly established and indeed, many of the structural features found in these complexes have become largely predictable. As a result of their widespread applicability in synthesis most of our understanding has come from detailed solution and solid state studies on lithium amides, though there has been a steady increase in the study of the heavier metal (Na, K) complexes.1,2 Limited hydrocarbon solubility, increased reactivity and possible reductions in selectivity are the reasons often quoted for the limited use of Na and K complexes, though implicit within this is also the belief that while structural differences will occur as a result of increasing cation size, the complex formed and the resulting chemistry will be essentially similar to that of the lithium counterpart. While most structural studies have indicated this is probably the case, there is also the possibility that the change in metal can effect dramatic and unexpected structural outcomes, which is important given that many alkali metal reagents are generated and used in situ.3
We reported recently that in the presence of pmdeta
(N,N,N′,N′,N″-pentamethylethy
lenetriamine), α-(methylbenzyl)benzylamido lithium will produce the
expected monomer,
(R)-[(Ph(Me)CH)(PhCH2)NLi·pmdeta],
1,4 while the sodium complex will
undergo the facile low-temperature transformation to a 2-azaallylic anion
system, {[Ph(Me)C![[horiz bar, double dot above]](https://www.rsc.org/images/entities/char_e0f2.gif) NC(H)Ph]Na·pmdeta}
2,6 as has also been described, in
detail, for the analogous alkali metal complexes of dibenzylamine.7 In comparing the Li complexes, the added methyl
group increased the stability of the amide complex to azaallyl formation.
We also noted the surprising paucity in solid and solution state structural
information which is available on enantiomerically pure chiral alkali metal
amides,4 especially in light of their
important role in synthesis.5 With this in
mind, we extended our studies to the complexes of
[R-(R*,R*)]-bis(α-m
ethylbenzyl)amine and herein report the solid state structure of
(R,R)-{[Ph(Me)CH]2NLi·pmdeta},
3, and the remarkable transformation of the amide to an enamide
with crystallisation of
{[PhC(CH2)NH]Na·pmdeta}24. Both structures have been determined by single crystal X-ray
diffraction.
NC(H)Ph]Na·pmdeta}
2,6 as has also been described, in
detail, for the analogous alkali metal complexes of dibenzylamine.7 In comparing the Li complexes, the added methyl
group increased the stability of the amide complex to azaallyl formation.
We also noted the surprising paucity in solid and solution state structural
information which is available on enantiomerically pure chiral alkali metal
amides,4 especially in light of their
important role in synthesis.5 With this in
mind, we extended our studies to the complexes of
[R-(R*,R*)]-bis(α-m
ethylbenzyl)amine and herein report the solid state structure of
(R,R)-{[Ph(Me)CH]2NLi·pmdeta},
3, and the remarkable transformation of the amide to an enamide
with crystallisation of
{[PhC(CH2)NH]Na·pmdeta}24. Both structures have been determined by single crystal X-ray
diffraction.
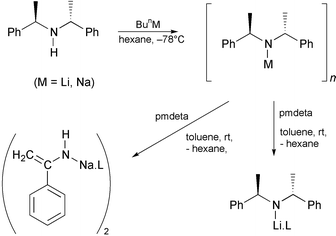
The preparative methods towards 3 and 4 are shown in Scheme 1. Complex 3 was prepared by the addition of BunLi to a hexane solution of (R,R)-bis(α-methylbenzyl)amine at −78 °C. Ligand pmdeta (1 equiv.) was added to the bright orange solution on reaching ambient temperature, resulting in a yellow precipitate which was redissolved on addition of toluene. Cooling the pale brown solution to −20 °C resulted in a large crop of pale yellow prismatic crystals, yield 67%.†
 | ||
| Scheme 1 | ||
Synthesis of 4 was by essentially the same method, the difference being that the amine was added to a hexane suspension of BunNa at −78 °C.† Pale yellow crystals of 4 were obtained at room temperature after ca. six days, but only after reduction of solvent to a minimum (ca. 5 ml for 5 mmol reaction).†
Complex 3 crystallises in the orthorhombic space group P212121 and is monomeric, as shown in Fig. 1.‡ In general, the structure is similar to that previously described for 1;4 the Li centre is four coordinate, bonding with the three available N atoms of pmdeta and Namido, is in a distorted tetrahedral environment and with comparable Li–N bonding distances. The most interesting feature of the structure though is the arrangement of the methyl groups, both in terms of their location in relation to the metal centre and their close proximity to the plane defined by C9, N1 and C1. Close H3C⋯Li interactions of 2.74 and 2.78 Å in the dimer (R,R)-{[Ph(Me)CH]2NLi·thf}2 are described as being of possible importance in explaining stereochemical outcomes in deprotonation reactions.8 Given the similarity in the conformations adopted by the amido moieties in the dimeric thf and monomeric pmdeta complexes of [(Ph(Me)CH)(PhCH2)NLi] it might have been expected that such close interactions and amide conformations would be carried through from (R,R)-{[Ph(Me)CH]2NLi·thf}2 into the structure of 3. As such, in viewing the solid state structure of 3 and the location of the Me groups it would be easy to conclude that the metal centre is dictating the orientation of the Ph(Me)CH moieties. However, while the asymmetry in Li⋯CH3 interactions observed in (R,R)-{[Ph(Me)CH]2- NLi·thf}2 remains, the analogous distances in 3 are significantly longer at 2.989 Å (C2–Li), and 3.325 Å (C10–Li). The closest distance is therefore only comparable with that observed in 1, of 3.04 Å. Also, the amido moieties in the thf and pmdeta structures of (R,R)-[Ph(Me)CH]2NLi do not adopt a similar orientation. In 3 the Me carbons and Namido are almost co-planar, which is evident if we consider a plane defined by N1, C9, C1 above which C10 and C2 lie 4.2 and 15.5°, respectively.
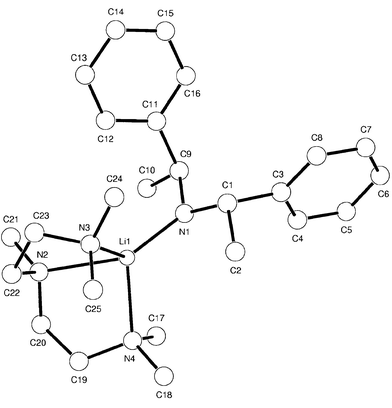 | ||
| Fig. 1 | ||
While the monomeric nature of 3 in the solid state was somewhat predictable the crystalline product and subsequent structure obtained from the Na reaction was not. We were probing whether the additional Me groups on the benzylic carbons would effect the formation of an azaallylic anion, as such the formation of the sodium enamide dimer, 4, was entirely unexpected.
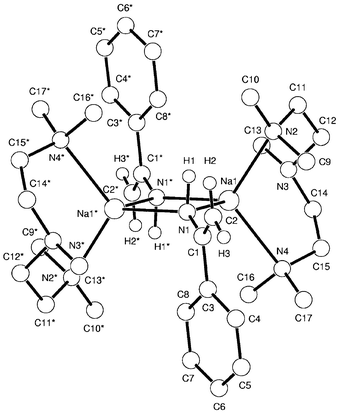
Complex 4 crystallises in the triclinic space group
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and is, as evident in Fig.
2, dimeric.‡ What is initially
striking about the complex is that it is an amide, with a formal M–N
bond, rather than a 1-azaallyl complex in which the Na cation is located
above the NCC fragment as observed in similar
systems,9 and which may have been
anticipated on metalation of a primary enamine. The propensity noted for
ketimides to undergo a 1, 3 sigmatropic rearrangement to 1-azallyl
complexes may hint at a possible mechanism for the formation of 4
involving the formation of [PhC(Me)NNa] and
PhC(H)CH2 as intermediates. Whether this involves the
formation and subsequent cleavage of a CN bond from a 2-azaallyl
intermediate complex is under investigation. A proton shift from Me onto N
as found in [{CH2C(tBu)N(H)-
Li·hmpa}2]10 with
subsequent stabilisation of the enamide over the 1-azallyl structure
via dimerisation in the solid state would result in 4.
The 1H and 13C NMR do not give a conclusive answer as
to whether the complex in solution adopts a 1-azaallyl arrangement, though
the chemical shift of the CH2 doublet, centred on
δ 2.65, is significantly upfield from the region expected
for PhCCH2 and is perhaps
indicative of a reduction in the double bond character.
and is, as evident in Fig.
2, dimeric.‡ What is initially
striking about the complex is that it is an amide, with a formal M–N
bond, rather than a 1-azaallyl complex in which the Na cation is located
above the NCC fragment as observed in similar
systems,9 and which may have been
anticipated on metalation of a primary enamine. The propensity noted for
ketimides to undergo a 1, 3 sigmatropic rearrangement to 1-azallyl
complexes may hint at a possible mechanism for the formation of 4
involving the formation of [PhC(Me)NNa] and
PhC(H)CH2 as intermediates. Whether this involves the
formation and subsequent cleavage of a CN bond from a 2-azaallyl
intermediate complex is under investigation. A proton shift from Me onto N
as found in [{CH2C(tBu)N(H)-
Li·hmpa}2]10 with
subsequent stabilisation of the enamide over the 1-azallyl structure
via dimerisation in the solid state would result in 4.
The 1H and 13C NMR do not give a conclusive answer as
to whether the complex in solution adopts a 1-azaallyl arrangement, though
the chemical shift of the CH2 doublet, centred on
δ 2.65, is significantly upfield from the region expected
for PhCCH2 and is perhaps
indicative of a reduction in the double bond character.
 | ||
| Fig. 2 | ||
The main features of 4 are the central cyclic (NNa)2 ring core about which the amide moieties adopt a trans configuration. This is wholly consistent the two other structurally characterised sodium primary amides with which 4 can be compared, [PhN(H)Na·pmdeta]25,11 and [2-PhOC4H6N(H)Na· pmdeta]2,12 and is a common feature of many sodium secondary amides.13 In all three cases the Na cation is five coordinate, however the tripodal connectivity of pmdeta in 5 makes it more closely related to 4. In 5 theanilino moieties tilt at an angle of 65° relative to the (NNa)2 ring plane, whereas in 4 the amide ligands are directly perpendicular, influenced no doubt by the sp2 nature of C1.
Acknowledgements
We thank the Australian Research Council and Monash University for financial support.Notes and references
- R. E. Mulvey, Chem. Soc. Rev., 1998, 27, 339 RSC; R. E. Mulvey, 1991, 20, 167..
- J. D. Smith, Adv. Organomet. Chem., 1999, 43, 267 Search PubMed; M. A. Beswick and D. S. Wright, Comprehensive Organometallic Chemistry II, ed. E. W. Abels, F. G. A. Stone and G. Wilkinson, Pergamon Press, New York, 1995, ch. 1; Search PubMed; E. Weiss, Angew. Chem., Int. Ed. Engl., 1993, 32, 1501 Search PubMed; F. S. Mair and R. Snaith, Dictionary of Organometallic Compounds, Chapman & Hall, London–New York, 1995, ch. 1; K. Gregory, P. v. R. Schleyer and R. Snaith, Adv. Inorg. Chem., 1991, 37, 47. CrossRef.
- C. Lambert and P. v. R. Scheleyer, Methods in Organic Chemistry, Thieme, Stuttgart, 1993, vol. 19d; Search PubMed; L. Brandsma, Preparative Polar Organometallic Chemistry, Springer-Verlag, Berlin–New York, 1987; Search PubMed; B. J. Wakefield, The Chemistry of Organolithium Compounds, Pergamon Press, Oxford, 1974, p. 204. Search PubMed.
- P. C. Andrews, P. J. Duggan, G. D. Fallon, T. D. McCarthy and A. C. Peatt, J. Chem. Soc., Dalton Trans., 2000, 1937 RSC.
- J. Busch-Peterson and E. J. Corey, Tetrahedron Lett., 2000, 41, 6941 CrossRef; K. Koga, Pure Appl. Chem., 1994, 66, 1487 CrossRef CAS; P. J. Cox, A. Persad and N. S. Simpkins, Synlett, 1992, 57, 5438; T. Honda, N. Kimura and M. Tsubuki, Tetrahedron: Asymmetry, 1993, 4, 21 CrossRef CAS; T. Honda, N. Kimura and M. Tsubuki, Tetrahedron: Asymmetry, 1993, 4, 1475 CrossRef CAS; K. Aoki, H. Naguchi, K. Koga and K. Tomioka, Tetrahedron Let., 1993, 34, 5105 CrossRef CAS; B. J. Bunn and N. S. Simpkins, J. Org. Chem., 1993, 58, 533 CrossRef CAS; P. J. Cox and N. S. Simpkins, Tetrahedron: Asymmetry, 1992, 2, 1 CrossRef CAS.
- P. C. Andrews, P. J. Duggan, G. D. Fallon, T. D. McCarthy and A. C. Peatt, J. Chem. Soc., Dalton Trans., 2000, 2505 RSC.
- P. C. Andrews, D. R. Armstrong, D. R. Baker, R. E. Mulvey, W. Clegg, L. Horsburgh, P. A. O’Neill and D. Reed, Organometallics, 1995, 14, 427 CrossRef CAS; P. C. Andrews, D. R. Armstrong, R. E. Mulvey and D. R. Reed, J. Organomet. Chem., 1990, 386, 287 CrossRef CAS.
- E. J. Edwards, S. Hockey, F. S. Mair, P. R. Raithby, R. Snaith and N. S. Simpkins, J. Org. Chem., 1993, 58, 6942 CrossRef.
- M. F. Lappert, J. Organomet. Chem., 2000, 600, 144 CrossRef CAS; A. Antiñolo, C. Huertas, I. del Hierro, M. F. Lappert, A. Otero, S. Prashar, A. M. Rodriguez and E. Villaseñor, Organometallics, 1998, 17, 5874 CrossRef CAS; B.-J. Deelman, M. F. Lappert, H.-K. Lee, T. C. W. Mak, W.-P. Leung and P.-R. Wei, Organometallics, 1997, 16, 1247 CrossRef CAS; M. F. Lappert and D.-S. Sui, J. Organomet. Chem., 1995, 500, 203 CrossRef CAS.
- D. R. Armstrong, W. Clegg, L. Dunbar, S. T. Liddle, M. MacGregor, R. E. Mulvey, D. Reed and S. Quinn, J. Chem. Soc., Dalton Trans., 1998, 3431 RSC.
- D. Barr, W. Clegg, L. Cowton, L. Horsburgh, F. M. MacKenzie and R. E. Mulvey, J. Chem. Soc., Chem. Commun., 1995, 891 RSC.
- I. Cragg-Hine, M. G. Davidson, A. J. Edwards, P. R. Raithby and R. Snaith, J. Chem. Soc., Dalton Trans., 1994, 2901 RSC.
- P. C. Andrews, N. D. R. Barnett, R. E. Mulvey, W. Clegg, P. A. O’Neil, D. Barr, L. Cowton, A. J. Dawson and B. J. Wakefield, J. Organomet. Chem., 1996, 518, 85 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: analytical data for 3 and 4. See http://www.rsc.org/suppdata/cc/b0/b007482i/ |
| ‡ Crystallographic
data: for 3: C25H41N4Li,
M = 404.56, T = 123 K, orthorhombic, space group
P212121, a = 9.987(2),
b = 13.692(3), c = 18.553(4) Å, V =
2537.0(9) Å3, Dc = 1.059 g
cm−3, Z = 4; F(000) = 888,
μ(Mo-Kα) = 0.64 cm−1,
2θmax = 56.9°, final R,
Rw = 0.091, 0.126. No =
3246 ‘observed’ [I > 2σ(I)]
reflections out of N = 5769 unique. GOF = 1.10. For 4: C13H31N4Na, M
= 266.4, T = 123 K, triclinic space group
P Crystallographic data collected on a Nonius Kappa CCD with crystals mounted under oil. All H atoms placed in calculated positions. CCDC 182/1835. See http://www.rsc.org/suppdata/cc/b0/b007482i/ for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2001 |