A molecular photoswitch based on an ‘axial-bonding’ type phosphorus(V) porphyrin
D. Raghunath
Reddy
and
Bhaskar G.
Maiya
*
School of Chemistry, University of Hyderabad, Hyderabad, 500 046, India.. E-mail: bgmsc@uohyd.ernet.in
First published on 15th December 2000
Abstract
Reversible isomerization of the two axial azobenzene subunits leads to modulation of the fluorescence due to the basal tetrapyrrolic chromophore in a new hexa-coordinated phosphorus(V) porphyrin 3, illustrating its utility as a molecular photoswitch.
‘Emitter–quencher’ assemblies based on porphyrin building blocks are attracting increasing attention because of their importance as either model compounds in photosynthetic research or photoswitches in the fabrication of molecular electronic/optical devices.1,2 While a great variety of covalently or non-covalently bound porphyrin–acceptor motifs are now known to closely mimic the initial, photoinduced electron transfer (PET) events of natural photosynthetic reactions,1 the utility of such motifs as photoswitches has not been firmly established. In this regard, it is interesting that a key recurring theme of most earlier attempts to construct porphyrin-based photoswitches involves intramolecular PET coupled to isomerization (E/Z) of the azobenzene group.3–8 This theme is an appealing one, and especially so, in view of the ready availability of PET-based non-porphyrinic photoswitches that incorporate azobenzene moieties in their architecture.2 However, barring a recent exception,4 photoswitching function has not been demonstrated in any of the porphyrin–azobenzene conjugates reported so far. Herein, we describe the luminescence on/off behavior observed in a novel, metalloid porphyrin-based photoswitch 3, which is constructed by utilizing the ‘axial-bonding’ capability of a phosphorus(V) porphyrin-(Fig. 1).
![Molecular structure of photoswitches investigated in the present study.
The numbers indicated adjacent to protons on the azobenzene moiety are the
corresponding porphyrin ring-current-induced 1H NMR
chemical shifts [i.e.
Δδ (free −
bound)] observed for 3E.](/image/article/2001/CC/b007784o/b007784o-f1.gif) | ||
| Fig. 1 Molecular structure of photoswitches investigated in the present study. The numbers indicated adjacent to protons on the azobenzene moiety are the corresponding porphyrin ring-current-induced 1H NMR chemical shifts [i.e. Δδ (free − bound)] observed for 3E. | ||
The hydroxide salt of photoswitch 3 was synthesized in 70% yield by reacting [5,10,15,20-tetra(tolyl)porphyrinato]phosphorus(V) dichloride 1 and 4-hydroxyazobenzene (excess) 2, in refluxing pyridine and purifying by column chromatography [silica gel, CHCl3–MeOH 10∶1, v/v)].9 The 1H NMR spectrum of this ‘axial-bonding’ type metalloid-porphyrin shows characteristic, porphyrin ring-current-induced upfield shifts for the protons on the axial aromatic ligands,10 with the magnitude of shift for a given proton being a function of its separation distance from the porphyrin π-plane (see Fig. 1). On the other hand, effects due to the substitution of axial chlorides by the aryloxo ligands are minimal for the porphyrin pyrrole-β and meso-aryl proton resonances. However, in the 31P NMR spectrum, the signal due to the central phosphorus ion of 3 was seen to be shifted downfield (δ −194.9, 85% H3PO4 external reference) compared to that of 1 (δ −229.4) but is within the typical range expected for hexa-coordinated diaryloxo phosphorus(V) porphyrins.9 Further support for the structural integrity of 3 arises from the appearance of its molecular ion peak at m/z = 1093 ([M]+) in the FAB mass spectrum.
The UV/VIS spectrum of 3 is essentially a summation of the spectra of 1 and 2 (1∶2 molar ratio), with the porphyrin Q- and B-bands [λmax/nm (log ε): 610 (4.03), 566 (4.28), 436 (5.36)] clearly distinguishable from the absorption due to the two trans azobenzene moieties in their E isomeric form [λmax/nm (log ε): 341 (4.86)] [Fig. 2 (top curve)]. These spectral features suggest that there is no electronic communication between the porphyrin and the azobenzene chromophores and, more importantly, that it is possible to individually address the photochemistry of these two subunits in this bichromophoric system. Accordingly, continuous irradiation of 3E (5.9 × 10−5 M, MeCN) at 345 ± 5 nm resulted in the time-dependent decrease of its absorption band centered at 341 nm concomitant with a slight increase of absorption in the B-band region, suggesting isomerization of the porphyrin-bound azobenzene subunits to produce 3Z.11 The reverse thermal reaction was also spectrally monitored and the E form could be recovered quantitatively (Fig. 2).
![Time-dependent UV/VIS spectral changes observed upon continuous
irradiation of 3E (solvent:
MeCN, [3E] = 5.9 ×
10−5 M). Top curve represents the initial spectrum with
the subsequent lower ones resulting from continuous irradiation of the
solution at 345 ± 5 nm at 25 °C for 1, 2, 5, 7, 9, 12 and 16
min, respectively (PTI 150 W Xe-arc lamp model A1010, PTI model 1366-MONO
monochromator). The dotted curve close to the top curve is the spectrum
obtained after keeping the irradiated solution in the dark for several
hours. Inset: fluorescence spectra of (––––)
unirradiated 3E, (–
–
–) 3Z obtained
upon irradiating 3E and of
(·
·
·
·)
3E resulting from the back
thermal reaction of 3Z
(solvent: MeCN; λexc = 465 nm).](/image/article/2001/CC/b007784o/b007784o-f2.gif) | ||
| Fig. 2 Time-dependent UV/VIS spectral changes observed upon continuous irradiation of 3E (solvent: MeCN, [3E] = 5.9 × 10−5 M). Top curve represents the initial spectrum with the subsequent lower ones resulting from continuous irradiation of the solution at 345 ± 5 nm at 25 °C for 1, 2, 5, 7, 9, 12 and 16 min, respectively (PTI 150 W Xe-arc lamp model A1010, PTI model 1366-MONO monochromator). The dotted curve close to the top curve is the spectrum obtained after keeping the irradiated solution in the dark for several hours. Inset: fluorescence spectra of (––––) unirradiated 3E, (– – –) 3Z obtained upon irradiating 3E and of (· · · ·) 3E resulting from the back thermal reaction of 3Z (solvent: MeCN; λexc = 465 nm). | ||
Excitation of a MeCN solution of 3E at 345 nm resulted in no fluorescence emanating from the azo chromophore as is the case with the precursor 2. On the other hand, the porphyrin component of the complex showed a fluorescence spectrum (λexc = 465/565 nm) typical of a hexa-coordinated phosphorus(V) porphyrin.9 The fluorescence quantum yield [Φf, estimated using (5,10,15,20-tetraphenylporphyrinato)zinc(II), ZnIITPP, as the standard] of 3E (0.01) is less than that of [5,10,15,20-tetra(tolyl)porphyrinato]phosphorus(V) dihydroxide {[PV(TTP)(OH)2]+, Φf = 0.045}. Interestingly, the fluorescence intensity due to 3Z (produced by continuous irradiation at 345 ± 5 nm) is only 60 ± 5% of that due to 3E, and the thermal back reaction regenerates the fluorescence of 3E as illustrated in Fig. 2 (inset). This E/Z interconversion was repeated 5–6 times with <5% loss of the material (UV/VIS and fluorescence), thus establishing the ability of 3 to be an effective and stable photoswitch.
What is the origin of weak fluorescence observed for 3 and what is the mechanism of its photoswitching function? Among the various possible mechanisms considered by us,† an intramolecular PET from the axial azobenzene donors to the singlet excited state of the basal phosphorus(V) porphyrin seems to be the most probable pathway for the quenching of fluorescence in 3E. This interpretation is consistent with not only the exoergicity for such a PET reaction (ΔGPET = −0.16 ± 0.03 eV‡ ), but also a similar interpretation made earlier for the quenching observed in a series of aryloxo phosphorus(V) porphyrins reported by us.9b Moreover, Φf for complex 4E (Fig. 1), which is endowed with the electron withdrawing nitro group at the axial azobenzene ligand, was seen (Φf = 0.035) to be more than that of 3E.§ Thus, accepting that PET is occurring between the axial ligand and the singlet porphyrin in this donor–acceptor complex, the photoswitching function demonstrated here can be rationalized in terms of the distance dependence of PET. As schematically represented in Fig. 3, the distance between the basal porphyrin and the axial ligand in 3Z is shorter than that in 3E explaining the additional fluorescence quenching observed for the former isomer.
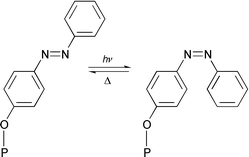 | ||
| Fig. 3 Reversible E/Z isomerization in 3 (P = porphyrin). | ||
Recently, electro-switch and proton-switch properties of an supramolecular ensemble comprising of ZnIITPP and axially ligated 4-(phenylazo)pyridine has been reported but, the effect of E/Z isomerization on the luminescence properties of this system was not observed.3 Similarly, studies on the covalently connected azobenzene–porphyrin conjugates reported by Hunter and Sarson revealed that photochemistry of the porphyrin components of these novel chromophoric assemblies is essentially unaltered but, the photochemical isomerization of their azobenzene components could not be detected.5 On the other hand, fluorescence properties of the early azobenzene–porphyrin systems reported by several groups were not investigated in detail.6–8 While this work was in progress, photoswitching features of an azobenzene-linked diporphyrin complex have been described.4 However, because of the extensive absorption by the two dissimilar porphyrin chromophores in the UV/VIS region, spectral detection of the E/Z isomerization in this system was not as facile as demonstrated here for 3.
In summary, the new phosphorus(V) porphyrin 3 is an effective and stable photoswitch. Its photoswitching ability is a result of the isomerization-induced modulation of the PET between the axial azobenzene subunits and the basal porphyrin scaffold. Further studies on 3 and other closely related ‘axial-bonding’ type photoswitches are currently in progress.
Acknowledgements
We thank the CSIR (New Delhi) for financial support of this work.Notes and references
- Reviews: T. Hayashi and H. Ogoshi, Chem. Soc. Rev., 1997, 26, 355 RSC; M. D. Ward, Chem. Soc. Rev., 1997, 26, 365 RSC; M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS.
- See, for example (and references therein): P. Belser, S. Bernhard, C. Blum, A. Beyeler, L. De Cola and V. Balzani, Coord. Chem. Rev., 1999, 190, 155 CrossRef; A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Soc. Rev., 1997, 26, 1515.
- J. Otsuki, K. Harada and K. Araki, Chem. Lett., 1999, 269 CrossRef CAS.
- S. Tsuchiya, J. Am. Chem. Soc., 1999, 121, 48 CrossRef CAS.
- C. A. Hunter and L. D. Sarson, Tetrahedron Lett., 1996, 37, 699 CrossRef CAS.
- M. Autret, M. le Plouzennec, C. Moinet and G. Simmonneaux, J. Chem. Soc., Chem. Commun., 1994, 1169 RSC.
- H. K. Hombrecher and K. Ludtke, Tetrahedron, 1993, 49, 9489 CrossRef CAS; H. K. Hombrecher, K. Ludtke and D. Koll, J. Prakt. Chem., 1996, 338, 257 CAS.
- K. H. Neumann and F. Vogtle, J. Chem. Soc., Chem. Commun., 1988, 520 RSC.
- (a) T. A. Rao and B. G. Maiya, J. Chem. Soc., Chem. Commun., 1995, 939 RSC; (b) T. A. Rao and B. G. Maiya, Inorg. Chem., 1996, 35, 4829 CrossRef CAS; (c) L. Giribabu, T. A. Tao and B. G. Maiya, Inorg. Chem., 1999, 38, 4971 CrossRef CAS.
- R. J. Abraham, G. R. Bedford, D. McNeille and B. Wright, Org. Mag. Reson., 1980, 14, 418 Search PubMed.
- S. Shinkai, T. Nakaji, T. Ogawa, K. Shigematsu and O. Manabe, J. Am. Chem. Soc., 1981, 103, 111 CrossRef.
Footnotes |
| † Excitation of 3 at 345 nm resulted in weak fluorescence in the range 550–750 nm but control experiments have suggested this to be entirely due to the residual absorption by the porphyrin chromophore. Moreover, the excitation spectrum of the compound (emission collected at the porphyrin fluorescence band maximum) did not show absorption corresponding to the azobenzene chromophore. In a separate set of experiments, neither the fluorescence of [P(TTP)(OH)2]+ was found to be quenched by compound 2 nor was any rate enhancement observed for the thermal back reaction of 3Z in the presence of externally added 2. Thus, fluorescence quenching observed here for 3 is not due to the energy transfer between the azobenzene and porphyrin subunits or the photochemical disscociation of azobenzene ligands. |
| ‡ Estimation of ΔGPET is based on the electrochemical redox potential data [Eox (axial ligand) = 1.52 V, Ered (basal porphyrin) = −0.35 V] and singlet energy of the porphyrin (2.03 eV9c). |
| § The presence of an electron withdrawing nitro group on the axial ligand is expected to decrease its donor capacity. Indeed, ΔGPET for 4 is close to 0.0 eV, consistent with its high Φf value. |
| This journal is © The Royal Society of Chemistry 2001 |