Molecular structure of and exchange coupling in a bis(semiquinone) complex
David A.
Shultz
*a,
Scot H.
Bodnar
a and
Jeff W.
Kampf
b
aDepartment of Chemistry, North Carolina State University, Raleigh, North Carolina 27695-8204, USA.. E-mail: David_Shultz@ncsu.edu
bDepartment of Chemistry, University of Michigan, Ann Arbor, Michigan 48109-1055, USA.. E-mail: jkampf@umich.edu
First published on 19th December 2000
Abstract
The first example of a structurally characterized, rationally designed, triplet ground-state bis(semiquinone) complex is presented; the intramolecular exchange coupling is ferromagnetic with J = +209 cm−1.
Molecular structures of organic biradical ligands are important to those concerned with construction of molecule-based magnetic materials using the metal-radical approach,1,2 and to those with an inherent interest in electronic properties of open-shell organic molecules.3
Recently, we reported several semiquinone-type ligands to be used as building blocks for high-spin molecules and open-shell materials.4,5 Among these new ligands is S = 1/2 quinonemethide-semiquinone (QMSQ) present in the complex (LZn)2(1′-H), the structure of which has been reported as well as its conversion to both a mixed-valent species and to a biradical, (LZn)21, shown below.5,6 Herein, we report both the molecular structure and magnetic properties of S = 1 (LZn)21 [TpCum,Me = hydrotris(3-cumenyl-5-methylpyrazolyl)borate].
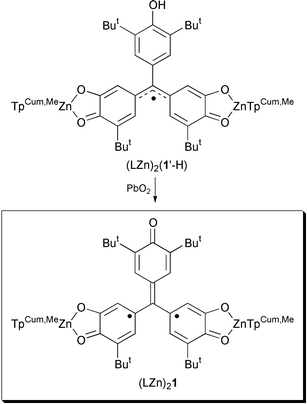
For clarity, we will refer to the mono-oxygenated rings of (LZn)2(1′-H) and (LZn)21 as A-rings, and the dioxygenated rings as either the dioxolene- or the B-rings. Structural changes that accompany conversion of (LZn)2(1′-H) to biradical (LZn)21 are consistent with the Lewis structures above.
The crystal structure of biradical (LZn)21 shows that the asymmetric unit contains two symmetry-independent molecules that differ primarily in torsion angles of A- and B-rings (and cumenyl ring torsions), and are therefore different conformers that also have an opposite sense of chirality.† An ORTEP of (LZn)21 is shown in Fig. 1, where hydrogens and cumenyl groups have been omitted for clarity. Bond lengths, angles, and torsions are summarized in Table 1 and Fig. 2.
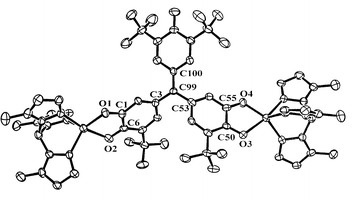 | ||
| Fig. 1 50% ORTEP of (LZn)21. Hydrogen atoms, cumenyl groups and THF molecules have been omitted for clarity. | ||
| Bond | R av/pm |
|---|---|
| a Averages for conformers I and II, the two symmetry-independent molecules in the asymmetric unit. b Torsion angles are relative to the plane made by atoms C3–C53–C99–C100. | |
| C1–O1, C55–O4 | 129.35, 128.35 |
| C6–O2, C50–O3 | 128.15, 127.15 |
| C1–C6, C50–C55 | 146.05, 146.55 |
| C3–C99, C53–C99 | 148.30, 146.95 |
| C99–C100 | 137.65 |
| Ring | Average torsion angles |
|---|---|
| C1–C6, C50–C55 | 50.33, 42.68 |
| C100–C105 | 18.05 |
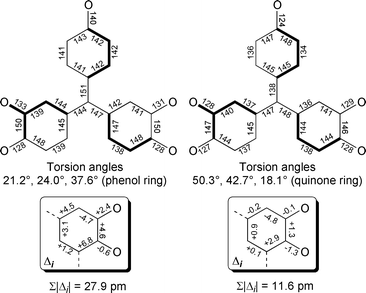 | ||
| Fig. 2 Comparison of structural features of (LZn)2(1′-H) (left) and (LZn)21 (right). Averaged bond lengths (in pm) are shown for (LZn)21. See text for details. | ||
The A-ring C–O bond of (LZn)21 (ca. 124 pm) is substantially shorter than the A-ring C–O single bond of (LZn)2(1′-H) (140 pm),5 consistent with carbonyl character for the former. In fact, as seen in Table 1 and Fig. 2, the pattern of long and short bond lengths for the A-ring in (LZn)21 confirms its quinoidal character.
The A-ring of (LZn)21 is twisted 18° while the dioxolene rings are twisted ca. 46° from the plane made by atoms C3, C53, C99 and C100, allowing conjugation of the dioxolene rings with the A-ring.
Fig. 2 shows bond length deviations (Σ|Δi |) of the B-rings from a 3,5-di-tert-butylorthosemiquinone ring.7,8 As we pointed out previously, the dioxolene rings of S = 1/2 (LZn)2(1′-H) are quite different from those of a semiquinone (Σ|Δi | = 27.9 pm) due to the QMSQ bond formulation.5 However, Σ|Δi| = 11.6 pm for the dioxolene rings of (LZn)21 indicating a greater similarity to a semiquinone, but also consistent with some delocalization of spin/charge into the A-ring, since we maintain that Σ|Δi | ≠ 0 is an indication of a delocalized electronic structure.
The magnetic susceptibility of microcrystalline (LZn)21 was measured from 2 to 300 K using a SQUID magnetometer, and is plotted as χparaTvs. T in Fig. 3. Modeling the temperature-dependent χparaT product of S = 1 molecules can be achieved by fitting to a modified Bleaney–Bowers expression:9,10
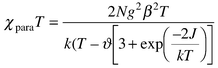 | (1) |
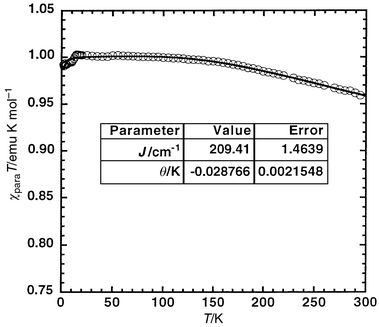 | ||
| Fig. 3 Temperature dependence of χparaT for biradical (LZn)21. | ||
The best fit of the data give J = +209 ± 1
cm−1 and ϑ
≈
−0.03 K. The very
small Weiss constant is consistent with negligible intermolecular
interactions due to the large, insulating hydrotrispyrazolylborate ligand,
while the moderately large, positive exchange parameter, J,
indicates that (LZn)21 is a triplet ground-state
biradical consistent with its π-connectivity. The symmetry of the
bis(semiquinone) ligand of (LZn)21 precludes degeneracy
of the SOMOs, nonetheless, delocalization of spin into the quinone-methide
unit (A-ring) gives rise to a substantial exchange integral, stabilizing
the triplet state relative to the singlet state. However, the different
spin distribution, lower symmetry, and therefore attenuated delocalization
into the A-ring results in a slightly smaller J for
(LZn)21 than for Yang’s biradical (J
![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) +245 cm−1).11,12
+245 cm−1).11,12
Acknowledgements
This work is funded by the National Science Foundation. D. A. S. thanks the Camille and Henry Dreyfus Foundation for a Camille Dreyfus Teacher-Scholar Award, and Professor Frank Tsui (Department of Physics, UNC-CH) for the use of his SQUID magnetometer.Notes and references
- A. Caneschi, D. Gatteschi, R. Sessoli and P. Rey, Acc. Chem. Res., 1989, 22, 392 CrossRef CAS
.
- S. Nakatsuji and H. Anzai, J. Mater. Chem., 1997, 7, 2161 RSC
- H. Bock, A. John and Z. Havlas, Angew. Chem., Int. Ed. Engl., 1993, 32, 416 CrossRef
- D. A. Shultz, A. K. Boal and N. P. Campbell, Inorg. Chem., 1998, 37, 1540 CrossRef CAS
- D. A. Shultz, S. H. Bodnar, R. K. Kumar and J. W. Kampf, J. Am. Chem. Soc., 1999, 121, 10664 CrossRef CAS
- D. A. Shultz and S. H. Bodnar, Inorg. Chem., 1999, 38, 591 CrossRef CAS
- C. G. Pierpont and R. M. Buchanan, Coord. Chem. Rev., 1981, 38, 45 CrossRef CAS
- C. G. Pierpont and C. W. Lange, Prog. Inorg. Chem., 1994, 41, 331 Search PubMed
- K. Inoue and H. Iwamura, Angew. Chem., Int. Ed. Engl., 1995, 34, 927 CrossRef CAS
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Sect. A, 1952, 214, 451 Search PubMed
- K. Mukai, K. Ishizu and Y. Deguchi, J. Phys. Soc. Jpn., 1969, 27, 783 Search PubMed
- P. W. Kopf and R. W. Kreilick, J. Am. Chem. Soc., 1969, 91, 6569 CrossRef CAS
Footnote |
| † Crystal data for (LZn)21: C126H160B2N12O8.25Z n2 which includes 3.25 molecules of THF solvate per Zn2 complex, red crystals, M = 2127.02, triclinic, space group P21/n; a = 38.110(4), b = 18.827(2), c = 38.448(4) Å, β = 118.546(2)°, V = 24233(4) Å3, Dc = 1.166 g cm−3, Z = 8, T = 158(2) K, R = 0.0730; wR2 = 0.2174, GOF = 1.016.Measurements were carried out on a standard Siemens SMART CCD-based X-ray diffractometer equipped with a normal focus Mo-target X-ray tube (λ = 0.71073 Å) operated at 2000 W power (50 kV, 40 mA). The structure was solved and refined with the Brüker SHELXTL (version 5.10) software package (G. M. Sheldrick, Bruker AXS, Madison, WI, 1997). All non-hydrogen atoms were refined anisotropically with the hydrogen atoms placed in idealized positions. Modest restraints were placed on the lattice solvates to retain chemically reasonable geometries. Refinement was carried out using blocked least-squares matrices. F(000) = 9088; reflections collected, 184229; no. of unique reflections, 34986; no. of parameters, 2655; R = 0.0730; wR2 = 0.2174; (Δρ)max = 1.967 e Å−3.CCDC 182/1860. See http://www.rsc.org/suppdata/cc/b0/b008037n/ for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2001 |