Porphyrins acting as external and internal ligands: preparation of conjugated trimetallic dimeric porphyrins
Sébastien
Richeter
,
Christophe
Jeandon
,
Romain
Ruppert
* and
Henry J.
Callot
*
Faculté de Chimie, 1 rue Blaise Pascal, 67008, Strasbourg, France.. E-mail: callot@chimie.u-strasbg.fr
First published on 18th December 2000
Abstract
A porphyrin bearing a peripheral enaminoketone function allows the preparation of conjugated dimeric porphyrins linked through metal ions.
Multiporphyrin assemblies are involved in several biological processes such as photosynthesis or electron transfer. Mimicking those assemblies and their activity has been a challenge for chemists and numerous examples of multiporphyrin compounds have been described in recent years.1–22 The connection between porphyrin nuclei may involve C–C bonds, hydrogen bonds, direct metal–metal or μ-X bonds, catenation of porphyrin ansa-derivatives, coordination of appropriate peripheral substituents to metal centers, etc. More recently, after the molecular wires described earlier by Crossley and Burn,8 several groups are pursuing efforts to obtain conjugated oligoporphyrinic molecules directly linked through one, two or even three covalent bonds.10–13 This covalent approach suffers some drawbacks: synthetic and separation problems, iterative reactions to obtain larger molecules, variations of the linking bonds limited to C–C or C–N.
The use of coordination bonds15–21 has the advantage of offering an additional choice: each metal center may present its own geometry and coordination number depending on the element and its oxidation level, in addition to the variations due to the nature of the internal coordination sites of the porphyrins.
Here we describe a substituted porphyrin acting both as an internal (with the four pyrrole nitrogens) and as an external (with an enaminoketone moiety) ligand. The external coordination site is coplanar and fully conjugated to the porphyrin aromatic system.22 In contrast to most systems assembled with metal ions, where little or no interaction is observed in the ground state between the porphyrins, the porphyrin–porphyrin conjugation in our dimers linked by metal ions will be illustrated by their electronic spectra and electrochemical behaviour.
meso-Tetraarylporphyrins bearing a keto group located on an additional ring attached to an aryl substituent and a pyrrolic β-carbon have been known since 1980 but their reactivity has been little investigated.23–26 In the course of a search for extended porphyrin chromophores we reacted ketone 1 with nitrogen nucleophiles. Under mild acid catalysis, reagents of the general composition H2N–X (X = OH, NHTs, OSO3H) gave in variable yield enaminoketone 2, the highest yield (90%) being observed with hydroxylamine O-sulfonic acid–AcOH–NaOAc in refluxing CH2Cl2. Enaminoketone 2a showed a UV–VIS similar to the starting material (λmax = 451, 568, 614 and 709 nm). The β-N–H NMR signals could be detected at δ 9.0 (N–H involved in a hydrogen bond with the carbonyl group) and δ 5.48 (free N–H, shielded by a vicinal phenyl ring).
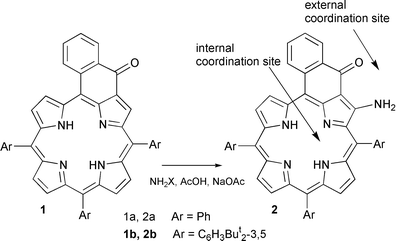
Enaminoketone 2 presents remarkable coordinating properties: in addition to the internal 4N tetradentate macrocyclic coordination site, the enaminoketone group is a potential N+O chelating external site (an analog of the acetylacetonate ligand) that might couple porphyrins to form homo- or hetero-polymetallic complexes. One would predict that the coordination at the enaminoketone site should be favoured over the introduction of metal ions in the core of the porphyrin, but one also expects the stability of the internal complex to be of orders of magnitude higher for the same metal ion; in particular with first row transition metals the porphyrin–metal–porphyrin connection is expected to be of low stability. Accordingly, heterometallation may be performed along two routes by using as an intermediate either an internal complex or the porphyrin base dimer (Scheme 1).
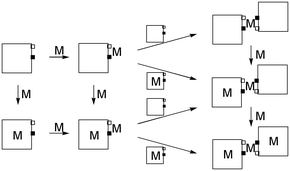 | ||
| Scheme 1 Metallation of enaminoketone 2 (squares): vertical arrows = metallation of internal site of porphyrin, slow and irreversible under the reaction conditions; horizontal arrows = metallation of external site (fast and possibly reversible reactions). | ||
Metallation of 2a (R = Ph) with nickel acetylacetonate (one or
more equiv.) rapidly gave a highly insoluble product containing one metal
and two porphyrins formulated as 3a (M1 = Ni;
M2 = M3 = H2). Treatment with acetic acid,
which is unable to remove nickel from the 4N site of a porphyrin,
quantitatively regenerated the starting material. However, on prolonged
reaction in refluxing toluene with a 1:1 porphyrin/nickel ratio,
metallation of the porphyrin occurred and gave mononuclear
Ni–2a, while excess reagent gave trinuclear
3a (M1 = M2 = M3 = Ni). The
formation of Ni–2a is best explained by the low
stability of the nickel bridge—it did not survive chromatographic
conditions—and the slow formation of the most stable metallation
product. The same trinuclear compound 3a (M1 =
M2 = M3 = Ni) was formed when
Ni–2a was treated with an excess of nickel
acetylacetonate. Similarly, several trimetallic bisporphyrins were prepared
(Ni/Cu/Ni; Ni/V![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O/Ni; Ni/Pd/Ni).
O/Ni; Ni/Pd/Ni).
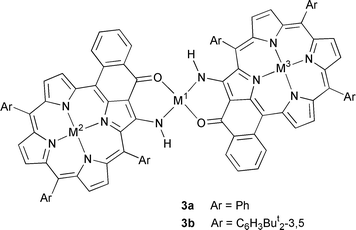
There remained the question of the relative arrangement of the porphyrins around the metal, since the dimers may have a center or a plane of symmetry. COSY and NOESY experiments confirmed the structure and the dimeric nature of 3. Additional information was obtained when we turned to the more soluble meso-tri(3,5-di-tert-butyl)phenylporphyrin series. Heating equimolecular amounts of palladium(II) acetate and enaminoketone 2b in refluxing toluene for 3 h led almost exclusively to 3b (M1 = Pd; M2 = M3 = H2). Again, NMR experiments (ROESY) confirmed the relative position of the porphyrins: HH ROESY cross signals were observed between two protons of the cyclised phenyl ring and the para proton and the tert-butyl protons of a meso-aryl group belonging to the other porphyrin. This compound survived chromatographic purification and is a good starting material for heterometallic dimer preparation. Indeed, metallation with zinc acetate gave the mono- and bis-metallated 3b (M1 = Pd; M2 = Zn; M3 = H2 or Zn).
These new porphyrin dimers all showed similar electronic spectra and an electrochemical behaviour indicative of the conjugation introduced through the connecting metal between the two aromatic rings. For example, the lowest energy band of 3a (M1–3 = Ni) shifts to 700 nm (ε = 33000 dm3 mol−1 cm−1) in comparison to the band observed at 649 nm (ε = 18500 dm3 mol−1 cm−1) for Ni–2a (Fig. 1). The HOMO–LUMO gap estimated from these data is reduced to 1.77 eV in the dimer 3a (M1–3 = Ni) compared to 1.91 eV for the monomer Ni–2a (already a very low value compared to the gap reported for nickel porphyrins, E ≈ 2.3 eV). The corresponding bases such as 3b (M1 = Pd; M2 = M3 = H2) display bands at even longer wavelengths (730 nm, ε = 39000 dm3 mol−1 cm−1), which can be compared with that of 2b (711 nm, ε= 8500 dm3 mol−1 cm−1).
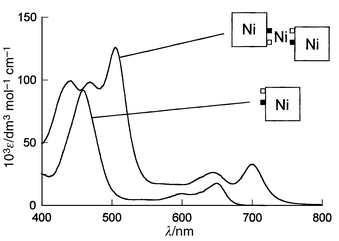 | ||
| Fig. 1 UV–VIS spectrum of 3a (M1–3 = Ni) and Ni–2a. | ||
The monomer Ni–2a showed two one-electron oxidations at 0.46 and 0.83 V vs. Fc/Fc+. In trimetallic 3a (M1–3 = Ni), the first oxidation is split into two one-electron oxidation waves (one electron per porphyrin) at 0.32 and 0.50 V, indicative of a strong interaction between the two porphyrins. The removal of the first electron therefore occurs at a potential 0.14 V lower than for the monomer (a value equal to the difference between the gaps taken from the electronic spectra). A two-electron oxidation (or rather twice a one-electron oxidation of each porphyrin ring) is further observed at 0.68 V. It seems that the removal of the first two electrons from the dimer has led to a disruption of the conjugation between the two rings. The dimer (already oxidised by two electrons) now behaves like two independant nickel monomers. All waves are quasi-reversible (ΔEp = 60–90 mV), showing that dimer 3a (M1–3 = Ni) remains intact during this overall four-electron oxidation–reduction process.
In conclusion, we confirmed that efficient porphyrin–porphyrin interaction could be achieved via metal coordination. The synthesis of bis-enaminoketoporphyrins where two external coordination sites are located in divergent directions is in progress. These compounds should allow the preparation of more highly conjugated oligomers.
Acknowledgements
We thank R. Graff for the NMR experiments.Notes and references
- J.-C. Chambron, V. Heitz and J.-P. Sauvage, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 6, p. 1 Search PubMed; J. K. M. Sanders, vol. 3, p. 347; S. Fukuzumi, vol. 8, p. 115; D. Gust and T. A. Moore, vol. 8, p. 153; J.-M. Barbe and R. Guilard, vol. 3, p. 211; J.-H. Chou, M. E. Kosal, H. S. Nalwa, N. A. Rakow and K. S. Suslick, vol. 6, p. 43..
- A. Osuka, N. Mataga and T. Okada, Pure Appl. Chem., 1997, 69, 797 CrossRef CAS.
- F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin III, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10 001 CrossRef CAS.
- A. K. Burrell and D. L. Officer, Synlett, 1998, 1297 CrossRef CAS.
- D. P. Arnold, Synlett, 1999, 296.
- M. G. H. Vicente, L. Jaquinod and K. M. Smith, Chem. Commun., 1999, 1771 RSC.
- H. L. Anderson, Chem. Commun., 1999, 2323 RSC.
- M. J. Crossley and P. L. Burn, J. Chem. Soc., Chem. Commun., 1987, 39 RSC; M. J. Crossley and P. L. Burn, J. Chem. Soc., Chem. Commun., 1991, 1569 RSC.
- W. J. Belcher, A. K. Burrell, W. M. Campbell, D. L. Officer, D. C. W. Reid and K. Y. Wild, Tetrahedron, 1999, 55, 2401 CrossRef CAS.
- L. Jaquinod, O. Siri, R. G. Khoury and K. M. Smith, Chem. Commun., 1998, 1261 RSC.
- R. G. Khoury, L. Jaquinod and K. M. Smith, Chem. Commun., 1997, 1057 RSC; N. Aratani, A. Osuka, Y. H. Kim, D. H. Jeong and D. Kim, Angew. Chem., Int. Ed. Engl., 2000, 39, 1458 CrossRef CAS.
- A. Tsuda, A. Nakano, H. Furuta, H. Yamochi and A. Osuka, Angew. Chem., Int. Ed. Engl., 2000, 39, 558 CrossRef CAS; K.-i. Sugiura, T. Matsumoto, S. Ohkouchi, Y. Naitoh, T. Kawai, Y. Takai, K. Ushiroda and Y. Sakata, Chem. Commun., 1999, 1957 RSC; I. M. Blake, L. H. Rees, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed. Engl., 2000, 39, 1818 CrossRef CAS.
- A. Tsuda, H. Furuta and A. Osuka, Angew. Chem., Int. Ed., 2000, 39, 2549 CrossRef CAS.
- R. Beavington and P. L. Burn, J. Chem. Soc., Perkin Trans. 1, 2000, 605 RSC.
- T. Imamura and K. Fukushima, Coord. Chem. Rev., 2000, 198, 133 CrossRef CAS.
- J. Wojaczynski and L. Latos-Grazynski, Coord. Chem. Rev., 2000, 204, 113 CrossRef CAS.
- A. Harriman, F. Odobel and J.-P. Sauvage, J. Am. Chem. Soc., 1995, 117, 9461 CrossRef CAS.
- M. J. Crossley, P. L. Burn, S. J. Langford and J. K. Prashar, J. Chem. Soc., Chem. Commun., 1995, 1921 RSC.
- T. A. Vannelli and T. B. Karpishin, Inorg. Chem., 1999, 38, 2246 CrossRef CAS.
- I. M. Dixon, J.-P. Collin, J.-P. Sauvage, F. Barigelletti and L. Flamigni, Angew. Chem., Int. Ed., 2000, 39, 1292 CrossRef CAS.
- Y. Diskin-Posner, S. Dahal and I. Goldberg, Angew. Chem., Int. Ed., 2000, 39, 1288 CrossRef CAS.
- A porphyrazine allowing the coordination of metal centers in the macrocyclic plane was recently described: N. Bellec, A. G. Montalban, D. B. G. Williams, A. S. Cook, M. E. Anderson, X. Feng, A. G. M. Barrett and B. M. Hoffman, J. Org. Chem., 2000, 65, 1774 CrossRef CAS.
- K. Henrick, P. G. Owston, R. Peters, P. A. Tasker and A. Dell, Inorg. Chim. Acta, 1980, 45, 161 CrossRef CAS.
- H. J. Callot, E. Schaeffer, R. Cromer and F. Metz, Tetrahedron, 1990, 46, 5253 CrossRef CAS.
- L. Barloy, D. Dolphin, D. Dupré and T. P. Wijesekera, J. Org. Chem., 1994, 59, 7976 CrossRef CAS.
- Y. V. Ishkov and Z. I. Zhilina, Zh. Org. Khim., 1995, 31, 136 Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |