AlCl3/ICl-Mediated iodo-carbocyclization of α-iodo cycloalkanones: a new entry to spirocyclic ketones
Chin-Kang Sha*, Fong-Cheng Lee and Hsien-Hsun Lin
Department of Chemistry, National Tsing Hua University, Hsinchu, 300, Taiwan, ROC.. E-mail: cksha@mx.nthu.edu.tw
First published on 11th December 2000
Abstract
Treatment of α-iodo cycloalkanones bearing an acetylenic side chain with AlCl3/ICl afforded spirocyclic ketones in good yields.
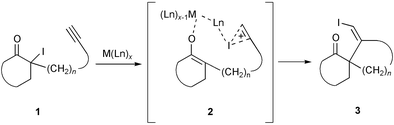
Spirocyclic systems are core skeletons of several important natural products, such as gloiosiphone A1 and ginkgolide B.2 They also constitute the main frameworks of spirocyclic chiral auxiliaries having a C2 axis of symmetry.3 During our work on radical cyclization of α-iodo ketones,4 we became interested in developing a general method for synthesis of spirocyclic ketones from α-iodo ketones. We envisaged that iodo-carbocyclization of α-iodo ketones, as depicted in Scheme 1, could be exploited for annulation of five- and six-membered rings. Generation of enolate from α-iodo ketone 1 with simultaneous transfer of I+ to the acetylenic moiety might be effected with a Lewis acid, M(Ln)x, to give intermediate 2. Subsequent cyclization of the intermediate 2 would afford spirocyclic ketone 3. In the past decade, free-radical atom-transfer cyclization of iodo substrates mediated with hexabutylditin5 or other reagents6 has emerged as a routine method. Ionic iodo-carbocyclization of iodo malonates7 and ionic seleno-carbocyclization of seleno ketones8 have also been described. In this communication, we report results obtained from our investigation of iodo-carbocyclization of α-iodo ketones.
 | ||
| Scheme 1 | ||
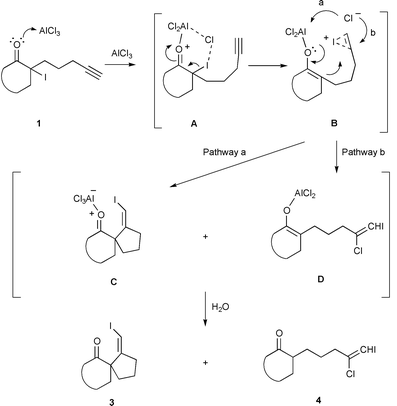
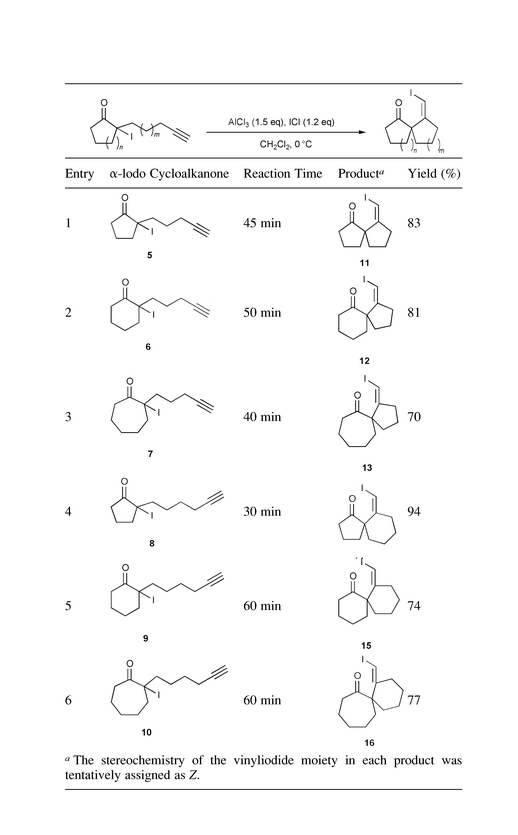
We first sought appropriate Lewis acids that could effect formation of an enolate from α-iodo ketones. Many Lewis acids including TiCl4, BCl3, AlMe3, Me2AlCl, SnCl4, MgBr2 and AlCl3 were examined. We found that AlCl3, Me2AlCl and TiCl4 effect the desired transformation of 1 to 3 in dichloromethane, although in low yield (10–20%). A plausible mechanism is proposed for the reaction using AlCl3 as catalyst, Scheme 2. AlCl3 reacts with α-iodo ketone to generate an aluminium enolate9 and ICl. The acetylenic moiety on the side chain then complexes with ICl to give intermediate B. Cyclization of B (pathway a) would afford AlCl3-complex C. In principle, AlCl3 is catalytic and gets regenerated at this stage. Because it would complex with the product, one equiv. of AlCl3 is needed. Upon aqueous work-up, complex C is hydrolyzed to spiro ketone 3. According to this mechanism, ICl is generated in the first step and participates in the subsequent cyclization. Therefore, we felt that addition of ICl from an external source might facilitate cyclization. Indeed, we found that treatment of iodo ketones 5–1010 with a mixture of AlCl3 (1.5 equiv.) and ICl (1.2 equiv.) in dichloromethane at 0 °C afforded spirocyclic products 11–16 in good yield.11 The results are summarized in Table 1. Products 11–16 are all obtained as a single geometric isomer and are tentatively assigned to be Z isomers.12 Presumably, the conformation of intermediate B, as depicted in Scheme 2, favors the formation of the exclusive Z isomers. Annulation of both five-membered rings (entries 1–3) and six-membered rings (entries 4–6) is achieved. In entries 1–3, by-product 4 is formed in trace amount (<5%) from direct addition of Cl− to the iodonium moiety, pathway b in Scheme 2.13 Since addition of an external source of ICl significantly enhances the yield of the carbocyclization process, an alternative mechanism involving enolate formation with simultaneous complexation of ICl to the acetylene unit is also possible.
 | ||
| Scheme 2 | ||
In conclusion, we have demonstrated that an ionic iodo-carbocyclization of α-iodo cycloalkanones can be effected with Lewis acid, AlCl3. Addition of ICl greatly enhances yields of spirocyclic ketones. In comparison with free-radical atom-transfer cyclization, the present method has two distinct advantages: (i) as tin reagents are not used, tedious separation of products from tin residues is avoided; (ii) whereas free-radical atom-transfer cyclization is only useful for synthesis of the five-membered ring, this method allows annulation of both five- and six-membered rings. Applications of this reaction for total synthesis of natural products are under investigation in our laboratory.
Acknowledgements
We thank the National Science Council of the Republic of China for financial support.Notes and references
- J. L. Chen, M. F. Moghaddam and W. H. Gerwick, J. Nat. Prod., 1993, 56, 1205 CrossRef CAS.
- N. Sakabe, S. Takada and K. Okabe, J. Chem. Soc., Chem. Commun., 1967, 259 RSC; K. Okabe, K. Yamada, S. Yamamura and S. Takada, J. Chem. Soc. C, 1967, 2201 RSC; K. Nakanishi, Pure Appl. Chem., 1967, 14, 89 CrossRef CAS.
- A. S. C. Chen, C.-C. Lin, J. Sun, W. Hu, Z. Li, W. Pan, A. Mi, Y. Jiang, T.-M. Huang, T.-K. Yang, J.-H. Chen, Y. Wang and G.-H. Lee, Tetrahedron: Asymmetry, 1995, 6, 2953 CrossRef; H. Suemune, K. Maeda, K. Kato and K. Sakai, J. Chem. Soc., Perkin Trans. 1, 1994, 3441 RSC; R. Brunner and H. Gerlach, Tetrahedron: Asymmetry, 1994, 5, 1613 CrossRef; J. A. Nieman, M. Parvez and B. A. Keay, Tetrahedron: Asymmetry, 1993, 4, 1973 CrossRef CAS.
- C.-K. Sha, T.-S. Jean and D.-C. Wang, Tetrahedron Lett., 1990, 31, 3745 CrossRef CAS; C.-K. Sha, R.-T. Chiu, C.-F. Yang, N.-T. Yao, W.-H. Tseng, F.-L. Liao and S.-L. Wang, J. Am. Chem. Soc., 1997, 119, 4130 CrossRef CAS; C.-K. Sha, K. C. Santhosh and S.-H. Lih, J. Org. Chem., 1998, 63, 2699 CrossRef CAS; C.-K. Sha and W.-Y. Ho, J. Chem. Soc., Chem. Commun., 1998, 2709 RSC; C.-K. Sha, F.-K. Lee and C.-J. Chang, J. Am. Chem. Soc., 1999, 121, 9875 CrossRef CAS.
- D. P. Curran and D. Kim, Tetrahedron Lett., 1986, 27, 5821 CrossRef CAS; D. P. Curran, Synthesis, 1988, 417 CrossRef CAS; D. P. Curran and C. Chang, J. Org. Chem., 1989, 54, 3140 CrossRef CAS; D. P. Curran, M.-H. Chen and D. Kim, J. Am. Chem. Soc., 1989, 111, 6265 CrossRef CAS; D. P. Curran and J. Tamine, J. Org. Chem., 1991, 56, 2746 CrossRef CAS.
- I. Marek and A. Chakraborty, J. Chem. Soc., Chem. Commun., 1999, 2375 RSC; I. Marek, J. Chem. Soc., Perkin Trans. 1, 1999, 535 RSC; K. Oshima, H. Yorimitsu, T. Nakamura and H. Shinokubo, J. Org. Chem., 1998, 63, 8604 CrossRef; D. P. Curran and C.-T. Chang, Tetrahedron Lett., 1990, 31, 933 CrossRef CAS.
- T. Taguchi, O. Kitagawa and T. Inoue, Tetrahedron Lett., 1992, 33, 2167 CrossRef; T. Taguchi, O. Kitagawa, T. Inoue and K. Hirano, J. Org. Chem., 1993, 58, 3106 CrossRef; T. Taguchi, O. Kitagawa and T. Inoue, Tetrahedron Lett., 1994, 35, 1059 CrossRef; T. Taguchi, T. Inoue, O. Kitagawa, S. Kurumizawa and O. Ochiai, Tetrahedron Lett., 1995, 36, 1479 CrossRef; T. Taguchi, O. Kitagawa, T. Suzuki, T. Inoue and Y. Watanabe, J. Org. Chem., 1998, 63, 9470 CrossRef; T. Taguchi, O. Kitagawa, T. Suzuki and H. Fujiwara, Tetrahedron Lett., 1999, 40, 2549 CrossRef; O. Kitagawa, H. Fujiwara, T. Suzuki, T. Taguchi and M. Shiro, J. Org. Chem., 2000, 65, 6819 CrossRef CAS.
- T. Toru, S. Kawai and Y. Ueno, Synlett, 1996, 539 CrossRef CAS.
- For the generation of aluminium enolate from α-bromo carbonyl compounds, see: S. Matsubara, N. Tsuboniwa, Y. Morizawa, K. Oshima and H. Zozaki, Bull. Chem. Soc. Jpn., 1984, 57, 3242 CAS.
- Iodo ketones 5–10 were prepared from the corresponding ketones according to our method: C.-K. Sha, J.-J. Young and T.-S. Jean, J. Org. Chem., 1987, 52, 3919 CrossRef CAS The required starting ketones were prepared by Yamashita’s procedure: T. Mino, S. Masuda, M. Nishio and M. Yamashita, J. Org. Chem., 1997, 62, 2633 CrossRef CAS.
- A representative procedure for iodo-carbocyclization: to a solution of compound 5 (200 mg, 0.72 mmol) in CH2Cl2 (7.2 mL) was added AlCl3 (150 mg, 1.08 mmol) at 0 °C. The mixture was stirred at 0 °C for 15 min, during which the color turned orange red. A solution of ICl in CH2Cl2 (1 M, 0.87 mL, 0.87 mmol) was added dropwise at 0 °C. The color became dark brown. The reaction mixture was stirred at 0 °C for 45 min and then quenched with H2O (20 mL), saturated Na2S2O3 solution (10 mL) and saturated NaHCO3 solution (10 mL). The mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine and dried (MgSO4). Concentration and silica gel column chromatography (hexane–EtOAc, 50∶1) gave product 11 (160 mg, 83%) as a pale yellow liquid. δH(300 MHz; CDCl3) 6.32 (s, 1H), 2.70−2.60 (m, 1H), 2.60−2.34 (m, 1H), 2.34−1.86 (m, 6H), 1.77−1.59 (m, 1H), 1,59−1.15 (m, 3H); δC(75 MHz; CDCl3) 211.2, 137.0, 73.4, 55.9, 40.7, 38.0, 34.4, 25.3, 19.5 (two carbons); IR (neat) 3069, 2953, 1732, 1602; MS (EI): m/z 277 (M + H+), 214 (13), 185 (52), 149 (31), 127 (47), 97 (45), 84 (100), 79 (34), 41 (55); HRMS (EI): Calc. for C10H14IO (M + H+) 277.0090. Found 277.0087..
- For purposes of comparison, E isomers of 11 and 13 were prepared from compounds 5 and 7 according to the photolytic hexabutylditin method (ref. 5). 1H NMR spectra of 11 and 13 were found to be different from those of E isomers. Therefore, all products 11–16 are tentatively assigned to be Z isomers..
- When the reactions of entries 1 and 2 were performed at −78 °C, products 11 and 12 were both obtained along with some by-product 4, in ratios of 1∶0.9 and 1∶0.8 respectively..
| This journal is © The Royal Society of Chemistry 2001 |