Synthesis of hydrolytically stable porphyrin C- and S-glycoconjugates in high yields†
Paolo
Pasetto
,
Xin
Chen
,
Charles Michael
Drain
* and
Richard W.
Franck
*
Department of Chemistry and Biochemistry, Hunter College and Graduate Center of The City University of New
York, 695 Park Avenue, New York, NY 10021, USA.. E-mail: cdrain@shiva.hunter.cuny.edu; rfranck@shiva.hunter.cuny.edu;; Tel: Phone: (212)650-3791;; Fax: (212)772-5332
First published on 14th December 2000
Abstract
Two C-glucosyl porphyrins are prepared using Ramberg–Backlund and Lindsey methods for the key conversions, and thiosugars are shown to react with perfluorophenylporphyrins.
Photofrin®, a mixture of hematoporphyrin oligomers, is currently used clinically for the photodynamic therapy (PDT) of cancers, but suffers from a variety of problems including solubility and dosing.1 Because of their potential selective binding to various cell types, porphyrins appended with a variety of saccharides have been examined as possible agents for PDT.1,2 Most recently, sugar-specific binding to rat hepatoma cells by porphyrin glycoconjugates has been described.2 Their efficacy as antibiotics and anti-viral agents is also under intense investigation.1 Since metalloporphyrins are well-established catalysts, the attachment of sugars to effect regio- and stereo-selective oxidations has also been examined, albeit with limited success.3 Additionally, there are several requirements for the successful commercialization of any of these adducts: (a) effective synthesis in high yields, (b) stable products, in this case to hydrolysis, and (c) highly pure compounds.
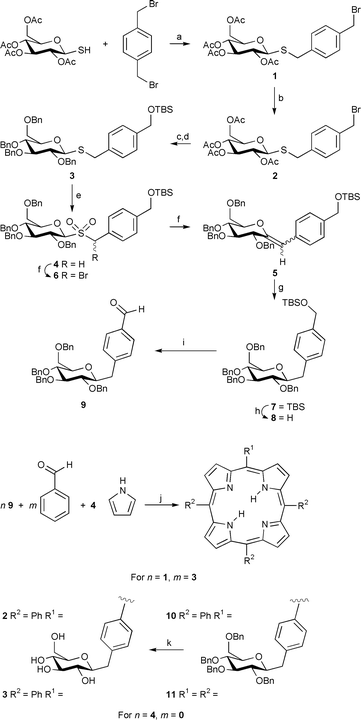 | ||
| Scheme 1 a, NaH, THF, rt, 2 h, 90%; b, TBDMSOH, Ag(OTf), 2,6-di-tert-butylpyridine, CH2Cl2, rt, 3 h, 60%; c, MeONa, MeOH, rt, 3 h; d, NaH, BnBr, Bu4N+I−, THF–DMF, rt, 8 h, 80% for 2 steps; e, MMPP, THF–EtOH–H2O, 60 °C, 2 h, 87%; f, CBr2F2, KOH 25% on alumina, CH2Cl2–t-BuOH, 0 °C to rt, 3 h, 88%; g, H2, Pd 5% on alumina, EtOAc, rt, 12 h, 95%; h, Bu4N+F−, THF, rt, 2 h, 98%; i, Oxalyl chloride, DMSO, CH2Cl2, −78 °C, then Et3N, −78 °C to rt, 1.5 h, 85%; j, BF3·OEt2, NaCl, rt, 5 h, then DDQ, rt, 30 min, 15% for 10, 53% for 11; k, H2, Pd 10% on carbon, EtOAc–MeOH, rt, 16 h, 98%. | ||
It is interesting that with few exceptions,4–6 the glycoconjugate has been oxygen linked via a glycosidic bond to a phenolic aryl porphyrin. When the syntheses of these O-linked materials are examined, one finds that disappointingly low yields are reported, considering that the Lindsey porphyrin synthesis yields are typically 45–65%.7 We believe that one problem may be that the frequently used O-glycosyloxybenzaldehyde starting materials, when subjected to the Lindsey porphyrin synthesis using BF3 catalysis, can undergo a competing and yield-reducing Suzuki O to C-glycoside rearrangement.8 Additionally, a perceived problem with the O-glycosides is the inherent possibility for glycosyl cleavage by acids, and in biological systems by glycohydrolases. We wish to describe our preliminary results, which provide possible solutions to both the issues of poor yields and hydrolytic instability, namely the preparation of C- and S-glycosylated aryl porphyrins in high yields. Both yield and stability are crucial factors for the successful incorporation of sugar moieties into porphyrin combinatorial libraries, since the linkage must be stable both during the formation, subsequent reactions, and in the presence of a variety of functional groups.
For the C-glycoside series, we chose to exploit earlier work from our laboratory where the Ramberg–Backlund synthesis is used to link sugars to aglycones.9,10 Thus, after screening several alternative functional equivalents of p-bromomethylbenzaldehyde, we settled on α,α′-dibromo-p-xylene as an inexpensive starting material. This was condensed with 1-thio-β-D-glucose tetraacetate11 to afford thioglucoside 1. Then conversion to the silylated material 2 was accomplished by treatment of 1 with tert-butyldimethylsilanol in the presence of silver triflate and 2,6-di-tert-butylpyridine.12 The replacement of the acetyl groups with benzyl ethers to obtain 3 was achieved by deprotection of 2 with sodium methoxide, followed by benzylation. The sulfide 3 was oxidized to the sulfone 4 with monoperoxyphthalic acid (MMPP). The resulting sulfone was employed in the Ramberg–Bäcklund synthesis of the exo-glucal 5 under Chan’s conditions.13 The two isomers (Z)-5 and (E)-5 in the ratio 8∶2 were identified by NOE measurements, which showed an effect between H-1 and H-3 in the case of the E-isomer. In some cases the intermediate α-bromosulfone 6 was isolated from the reaction mixture and then converted to the exo-glucal 5 by treatment with base. The hydrogenation of 5 with palladium 5% on alumina afforded the β-C-glucoside 7, identified by the coupling constant J = 9.2 Hz, indicating an anti-diaxial configuration of the anomeric H-2 with respect to H-3. The cleavage of the silyl ether 7, followed by Swern14 oxidation gave the aldehyde 9, which was utilized in the synthesis of the porphyrins 10 and 11 in 15 and 53% yields respectively, under Lindsey conditions.7 The materials obtained showed the expected spectroscopic data. The hydrogenolysis of the benzylic protecting groups yielded the porphyrins 12 and 13, without reduction of the porphyrin. S-Glycosides were chosen as worthy conjugates because they are considered to be good mimics of O-glycosides with enhanced stability toward enzymatic hydrolysis.15
The second reaction sequence begins with 5,10,15,20-tetra(perfluorophenyl)porphyrin which can be routinely made in gram quantities in high yields by the Lindsey procedure.7 The reactivity of the para-fluoro group toward a large variety of nucleophilic substitution reactions has been well established.4,16 The tetrakis(thiogalactosyl) and tetrakis(thioglucosyl) derivatives of this fluorinated porphyrin are formed in >85 and 90% yields, respectively, by modifications of previously described procedures.16 Specifically, the porphyrin is dissolved in amine-free DMF, five equiv. of the sodium salt of the thiosugar added, and the mixture stirred at rt for 4 h. Purification is accomplished on silica-gel using an ethanol–ethyl acetate gradient as eluent.
DNA photocleavage assays are widely used to evaluate the photoreactivity of porphyrin compounds, though DNA is not the primary site of Photofrin activity.1,5,6 Thus we used standard plasmid photocleavage assays to evaluate the photoreactivity and compare these results with those for other porphyrins. The conditions used to evaluate the photodamage to plasmid DNA caused by porphyrin compounds varies, but our analysis (see Table 1 indicates the compounds reported herein exhibit poor photoreactivity which is consistent with other glycosylated porphyrins.1,5,6 The amphipathic character of the macrocycle derivative has been shown to correlate to cell toxicity in vitro,1 so the partition coefficients are reported. To date, for this class of compounds, the activity is much poorer than the 5,10- bis(4-methylpyridinium)-15-(4-methylphenyl)-20-(4-hydroxy- phenyl)porphyrin (DiMePy+MeOHP) found by combinatorial methods,17 and for some other tetraphenylporphyrin derivatives.1
In conclusion, both the C and S linked glycoporphyrins can be synthesized in high yields exceeding 50% based on starting aldehydes using more efficient synthetic strategies. These compounds are stable to hydrolysis and show some photoactivity. Despite significant efforts by a large number of groups, there are few effective photodynamic therapy (PDT) agents, thus there remains a need to further develop the chemistry and the structure–activity relationships of this class of compounds. In contrast to the present clinical use of Photofrin® for the PDT of cancers, it may be found that different porphyrinic compounds are needed to more effectively treat different cancerous tissues, or for other therapeutic uses.
Acknowledgements
The authors thank Margareta Sorenson and Dr Clifford Soll, and acknowledge the support of NIGMS SCORE Program 1S06GM60654, NSF CAREER 9732950 to C. M. D.; PSC-CUNY awards to C. M. D. and R. W. F.; NIH GM51216 to R. W. F. Chemistry department infrastructure is partially supported by RCMI grant NIH RR03037. R. W. F. dedicates this article to Professor Steven Weinreb on the occasion of his 60th birthday. ®QLT Phototherapeutics Inc.Notes and references
- R. Bonnett, P. Charlesworth, B. D. Djelal, S. Foley, D. J. McGarvey and T. G. Truscott, J. Chem. Soc., 1999, 325 Search PubMed; E. D. Sternberg, C. Bruckner and D. Dolphin, Tetrahedron, 1998, 54, 4151 CrossRef CAS; T. J. Dougherty and D. Kessel, Rev. Contemp. Pharmacother., 1999, 10, 19 Search PubMed; B. R. Munson and R. J. Fiel, Nucleic Acids Res., 1992, 20, 1315 CAS.
- K. Fujimoto, T. Miyata and Y. Aoyama, J. Am. Chem. Soc., 2000, 122, 3558 CrossRef CAS.
- P. Maillard, J. L. Guerquin-Kern and M. Momenteau, Tetrahedron Lett., 1991, 32, 4901 CrossRef CAS.
- A thioether link between tetra(tetrafluorophenyl)porphyrin and C-6 of glucose embedded in cyclodextrin has been reported: J. Yang and R. Breslow, Angew. Chem., Int. Ed., 2000, 39, 2692, and references therein. Search PubMed.
- C-glycosides: M. Cornia, M. Menozzi, E. Ragg, S. Mazzini, A. Scarafoni, F. Zanardi and G. Casiraghi, Tetrahedron, 2000, 56, 3977, and references therein; Search PubMed; M. Momenteau, P. Maillard, M.-A. De Bélinay, D. Carrez and A. Croisy, J. Biomed. Opt., 1999, 4, 298 Search PubMed; N. Ono, M. Baugauchi and K. Maruyama, Tetrahedron Lett., 1992, 33, 1629 CrossRef CAS.
- S-glycosides linked to alkyl groups: I. Sylvain, R. Benhaddou, V. Carre, S. Cottaz, H. Driguez, R. Granet, M. Guilloton and P. Krausz, J. Porphyrins Phthalocyanines, 1999, 3, 1 and references therein. Search PubMed.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827 CrossRef CAS; F. Li, K. Yang, J. S. Tyhonas, K. A. MacCrum and J. S. Lindsey, Tetrahedron, 1997, 53, 12339 CrossRef CAS.
- T. Matsumoto, T. Hosoya and K. Suzuki, Synlett, 1991, 709 CrossRef CAS; T. Matsumoto, T. Hosoya and K. Suzuki, Tetrahedron Lett., 1990, 31, 4629 CrossRef CAS.
- P. S. Belica and R. W. Franck, Tetrahedron Lett., 1998, 39, 8225 CrossRef CAS; G. Yang, R. W. Franck, H.-S. Byun, R. Bittman, P. Samadder and G. Arthur, Org. Lett., 1999, 1, 2149 CrossRef CAS.
- F. K. Griffin, P. V. Murphy, D. E. Patterson and R. J. K. Taylor, Tetrahedron Lett., 1998, 39, 8179 CrossRef CAS; M-.L. Alcaraz, F. K. Griffin, D. E. Patterson and R. J. K. Taylor, Tetrahedron Lett., 1998, 39, 8183 CrossRef CAS; R. J. K. Taylor, F. K. Griffin and D. E. Paterson, Angew. Chem., Int. Ed., 1999, 38, 2939 CrossRef; A. D. Campbell, D. E. Paterson, T. M. Raynham and R. J. K. Taylor, Chem. Commun., 1999, 1599 RSC.
- D. Horton, in Methods in Carbohydrate Chemistry, eds. R. L. Whistler and M. L. Wolfrom, Academic Press Inc., New York, 1963, Vol. II, p. 433–437. Search PubMed.
- R. M. Burk, T. S. Gac and M. B. Roof, Tetrahedron Lett., 1994, 35, 8111 CrossRef CAS.
- T. L Chan, S. Fong, Y. Li, T. O. Man and C. D. Poon, Chem. Commun., 1994, 1771 RSC.
- K. Omura and D. Swern, Tetrahedron, 1978, 34, 1651 CrossRef CAS.
- B. D. Johnston and B. M. Pinto, J. Org. Chem., 2000, 65, 4607, and references therein Search PubMed; J. C. Wilson, M. J. Kiefel, D. I. Angus and M. von Itzstein, Org. Lett., 1999, 1, 443; for recent examples of thioglycoside stability toward a glycohydrolase. Search PubMed.
- S. J. Shaw, K. J. Elgie, C. Edwards and R. W. Boyle, Tetrahedron Lett., 1999, 1595 CrossRef CAS.
- C. M. Drain, X. Gong, V. Ruta, C. E. Soll and P. F. Chicoineau, J. Comb. Chem., 1999, 1, 286 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details. See http://www.rsc.org/suppdata/cc/b0/b008489l/ |
| This journal is © The Royal Society of Chemistry 2001 |