Structural studies on dicopper(II) compounds with catechol oxidase activity†
Heidi
Börzel
,
Peter
Comba
* and
Hans
Pritzkow
Universität Heidelberg, Anorganisch-Chemisches Institut, INF 270, D-69120, Heidelberg, Germany.. E-mail: comba@akcomba.aci.uni-heidelberg.de
First published on 19th December 2000
Abstract
The X-ray crystal structures of three low molecular weight models of catechol oxidase with three different coordination modes are reported; and the compound with a bridging catecholate is shown to be the catalytically most active form.
A range of biological dicopper sites have similar structures, with three histidine donors for each of the two Cu sites and Cu–Cu distances of ca. 3.5 Å. These include the oxygen transport protein hemocyanin, the oxygenation enzyme tyrosinase and the oxidation enzyme catechol oxidase. The active sites of these dicopper proteins are structurally well characterized;1–6 thorough spectroscopic studies, combined with computational investigations, have defined the electronic structures of the active sites,7,8 and extensive kinetic studies9,10 have led to the proposal of mechanisms for oxygen transport and activation, as well as oxygen and electron transfer. The assumption of a bridging catecholate as the active species in catechol oxidase7 was recently challenged on the basis of crystallographic data, which suggested an active state with a monodentate catecholate.6
Low molecular weight model compounds have helped to understand structural, electronic and mechanistic features and are expected to be useful for the development of new catalysts. A number of CuII-based models with catechol oxidase activity have been reported,11–16 but only few relevant experimental structures with coordinated catecholate have appeared so far.11 In particular, there is no example where the various coordination modes of catecholate, including the putative intermediates with bridging or monodentate catecholate, have been analyzed with identical coligands, and for some relevant coordination models there have not been any structural data available so far.
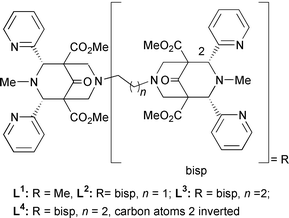
We have successfully used mono- and di-nuclear CuI and CuII compounds with bispidine-type ligands to stabilize μ-peroxodicopper(II) compounds.17,18 We now present preliminary results on the catechol oxidase activity of the corresponding CuII compounds with 3,5-dtbc in MeOH (3,5-dtbc = 3,5-di-tert-butylcatechol; spectrophotometric analysis of the o-quinone product).14,15 The mononuclear CuII complex of L1 is inactive, in contrast to the dinuclear compounds with L2 and L3. One equivalent of [Cu2(L3)(solv)2]4+ (solv = solvent) produces in a stoichiometric process 2 equivalents of quinone, while 13 equivalents of quinone are produced per h in a catalytic reaction with [Cu2(L2)(solv)2]4+. It emerges that the catalytic activity is a function of the catechol binding mode and stability, and this may differ for all three CuII compounds.
To examine this, the electronically deactivated substrate tccH2 (tccH2 = tetrachlorocatechol) was added in various concentrations to methanolic solutions of the three CuII compounds, and substrate binding was monitored spectrophotometrically. For [Cu(L1)(solv)]2+ a strong absorption band appeared at ca. 450 nm; for [Cu2(L2)(solv)2]4+ and [Cu2- (L3)(solv)2]4+ equilibria between species with absorptions at ca. 450 nm and ca. 530 nm were established; with L2 the species with the lower energy transition was more stable than with L3, where it disappeared with an excess of catechol (see ESI†). These results are in accord with the assumption that [Cu2- (L2)(solv)2]4+ and [Cu2(L3)(solv)2]4+ lead to catecholate-bridged active compounds, while [Cu(L1)(solv)]2+ leads to a mononuclear catecholate compound. A molecular model19 (see Fig. 1) indicates that the ethylene-bridged dicopper(II) compound is suitable and highly preorganized for a bridging catecholate.
![Molecular model of
[Cu2(L2)(tcc)]2+.](/image/article/2001/CC/b008714i/b008714i-f1.gif) | ||
| Fig. 1 Molecular model of [Cu2(L2)(tcc)]2+. | ||
Single crystals of [Cu(L1)(tccH)](ClO4) 1, [Cu(L1)(tcc)] 2 and [Cu2(L3)(tcc)](ClO4)23 were obtained by reaction of the CuI precursors with the fully chlorinated quinone (tcbq) or with tccH2 and O2, followed by slow evaporation of the solvent (full experimental details, including synthetic procedures, crystal growth, spectroscopic (IR, UV–VIS) and elemental analytical data, are given as ESI†).20,21 ORTEP plots are shown in Fig. 2. The structure of 1 is of poor quality, but catecholate binds unambiguously as a monodentate, monoprotonated ligand (see analytical data in the ESI†). Also shown in Fig. 2 is an ORTEP plot of [Cu2(L4)(tcc)2] 4. L4 has a non-coordinating pyridyl substituent at each copper center and, therefore, leads to copper(II) chromophores with the usual in-plane chelating catecholate coordination mode.
![ORTEP plots of [Cu(L1)(tccH)]+1,
[Cu(L1)(tcc)] 2,
[Cu2(L3)(tcc)]2+3 and
[Cu2(L4)(tcc)2] 4 (50%)
probability level). H-atoms, ester groups and counter ions have been
omitted for clarity. Selected bond lengths (Å) and angles (°) for
1; 2; 3; 4; Cu(1)–N(1):
2.020(11); 2.042(2); 2.027(5); 2.094(5). Cu(1)–N(2): 2.320(12);
2.433(2); 2.359(5); 2.293(5). Cu(1)–N(3) 1.987(11); 2.009(2);
2.004(6); —. Cu(1)–N(4) 1.989(11); 2.031(2); 1.985(6);
1.995(5). Cu(1)–O(7) 1.915(9); 1.909(2); 1.898(4); 1.947(4).
Cu(1)⋯O(8) 2.76; 2.46; —; 1.898(4). N(1)–Cu(1)–N(2)
84.06(44); 80.97(8); 83.93(19); 82.24(17). N(1)–Cu(1)–N(3)
81.71(44); 80.56(9); 81.5(2); —. N(1)–Cu(1)–N(4)
81.94(44); 82.15(9); 82.4(2); 80.37(18). N(1)–Cu(1)–O(7)
176.85(47); 172.97(8); 178.1(2); 164.10(18). N(2)–Cu(1)–N(3)
96.66(44); 91.42(8) 95.6(2); —. N(3)–Cu(1)–N(4)
161.28(48); 161.58(9); 161.7(2); —. 4:
O(8)–Cu(1)–O(7) 86.70(17).](/image/article/2001/CC/b008714i/b008714i-f2.gif) | ||
| Fig. 2 ORTEP plots of [Cu(L1)(tccH)]+1, [Cu(L1)(tcc)] 2, [Cu2(L3)(tcc)]2+3 and [Cu2(L4)(tcc)2] 4 (50%) probability level). H-atoms, ester groups and counter ions have been omitted for clarity. Selected bond lengths (Å) and angles (°) for 1; 2; 3; 4; Cu(1)–N(1): 2.020(11); 2.042(2); 2.027(5); 2.094(5). Cu(1)–N(2): 2.320(12); 2.433(2); 2.359(5); 2.293(5). Cu(1)–N(3) 1.987(11); 2.009(2); 2.004(6); —. Cu(1)–N(4) 1.989(11); 2.031(2); 1.985(6); 1.995(5). Cu(1)–O(7) 1.915(9); 1.909(2); 1.898(4); 1.947(4). Cu(1)⋯O(8) 2.76; 2.46; —; 1.898(4). N(1)–Cu(1)–N(2) 84.06(44); 80.97(8); 83.93(19); 82.24(17). N(1)–Cu(1)–N(3) 81.71(44); 80.56(9); 81.5(2); —. N(1)–Cu(1)–N(4) 81.94(44); 82.15(9); 82.4(2); 80.37(18). N(1)–Cu(1)–O(7) 176.85(47); 172.97(8); 178.1(2); 164.10(18). N(2)–Cu(1)–N(3) 96.66(44); 91.42(8) 95.6(2); —. N(3)–Cu(1)–N(4) 161.28(48); 161.58(9); 161.7(2); —. 4: O(8)–Cu(1)–O(7) 86.70(17). | ||
The C–C and C–O bond lengths of tcc2− and tccH− confirm the assignment as coordinated catecholate in all four structures. The geometry around the CuII centers can be described as square pyramidal, with N1, the pyridine N-atoms and one of the catecholate O atoms in the square plane and N2 at the axial position; in 2 O8 completes the coordination sphere to an elongated octahedron, and in 4 the second catecholate-O substitutes one of the pyridine-N atoms. 2 is only sparingly soluble in most solvents; the UV–VIS spectrum of a very dilute solution (MeCN) indicates an equilibrium between the mono- and bidentate coordination modes of catecholate, i.e. structures 1 and 2 (Fig. 2).
The most prominent structural difference between 3 and the other known structure of a catecholate-bridged dicopper(II) complex is the orientation of the catecholate bridge (angle between the line through the two metal ions and the line through the two catecholate O-atoms: 13.6° in 3, 32.0° in [Cu2(L2)(TCC)]2+ (computed), 63.1° in11). It is interesting that the increasing puckering of the catecholate bridge correlates with the catalytic activity in the 3,5-dtbc to 3,5-dtbq reaction.
For the model reactions involving bispidine-based ligands it appears that catecholate oxidation occurs at a catecholate-bridged dicopper(II) site by electron transfer from the catechole to the CuII ions; reoxidation of the Cu centers by molecular oxygen produces water and the active catalyst (the absence of H2O2 has been checked by reaction with KI). An interesting question is how thermally stable copper(II) peroxo compounds, generated during the catalytic process, affect the reaction. This and a thorough analysis of the electronic structures of the various structural modes are the subject of further studies in this area.
Generous financial support by the German Science Foundation (DFG), the Fonds of the Chemical Industry (FCI) and the Landesgraduiertenförderungsprogramm of Baden-Württemberg (fellowship to H. B.) is gratefully acknowledged.
Notes and references
- W. P. J. Gaykema, W. G. J. Hol, J. M. Vereijken, N. M. Soeter, H. J. Bak and J. J. Beintema, Nature, 1984, 309, 23 CrossRef CAS.
- A. Volbeda and W. G. J. Hol, J. Mol. Biol., 1989, 209, 249 CAS.
- B. Hazes, K. A. Magnus, C. Bonaventura, J. Bonaventura, Z. Dauter, K. H. Kalk and W. G. J. Hol, Protein Sci., 1993, 2, 597 CAS.
- K. A. Magnus, B. Hazes, H. Ton-That, C. Bonaventura, J. Bonaventura and W. G. J. Hol, Proteins, 1994, 19, 302 CAS.
- M. E. Cuff, K. I. Miller, K. E. van Holde and W. A. Hendrickson, J. Mol. Biol., 1998, 278, 855 CrossRef CAS.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563 CrossRef CAS.
- H. Decker, R. Dillinger and F. Tuczek, Angew. Chem., 2000, 112, 1656 CrossRef.
- B. Salvato, M. Santamaria, M. Beltramini, G. Alzuet and L. Casella, Biochemistry, 1998, 37, 14065 CrossRef CAS.
- K. D. Karlin, S. Kaderli and A. D. Zuberbühler, Acc. Chem. Res., 1997, 30, 139 CrossRef CAS.
- K. D. Karlin, Y. Gultneh, T. Nicholson and J. Zubieta, Inorg. Chem., 1985, 24, 3725 CrossRef CAS.
- M. R. Malachowski, H. B. Huynh, L. J. Tomlinson, R. S. Kelly and J. W. Furbee jun., J. Chem. Soc., Dalton Trans., 1995, 31 RSC.
- J. Manzur, A. M. Garcia, R. Rivas, A. M. Atria, J. Valenzuela and E. Spodine, Polyhedron, 1997, 16, 2299 CrossRef CAS.
- J. Reim and B. Krebs, J. Chem. Soc., Dalton Trans., 1997, 3793 RSC.
- E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553 CrossRef CAS.
- E. Monzani, G. Battaini, A. Perotti, L. Casella, M. Gullotti, L. Santagostini, G. Nardin, L. Randaccio, S. Geremia, P. Zanello and G. Opromolla, Inorg. Chem., 1999, 38, 5359 CrossRef CAS.
- H. Börzel, P. Comba, C. Katsichtis, W. Kiefer, A. Lienke, V. Nagel and H. Pritzkow, Chem. Eur. J., 1999, 5, 1716 CrossRef CAS.
- H. Börzel, P. Comba, K. S. Hagen, C. Katsichtis and H. Pritzkow, Chem. Eur. J., 2000, 6, 914 CrossRef CAS.
- P. Comba, T. W. Hambley, N. Okon and G. Lauer, ‘MOMEC97 a molecular modeling package for inorganic compounds’ CVS, Softwareentwicklung, e-mail:cvs@t-online.de, 1997. The force field is that used before for the corresponding peroxo compounds.17 Structural parameters involving the catecholate bridge were constrained to values derived from the two known structures, see this work and ref. 11..
-
Crystal structure determination: data were collected at
−100 °C with a Bruker-AXS CCD diffractometer (Mo-Kα
radiation, λ = 0.71073 Å, ω-scans).
Structures were solved by direct methods and refined against
F2 (SHELXTL V5.10). 2:
C31H31N5O8Cl4Cu,
M = 806.9, monoclinic P21/c,
a = 16.5741(9), b = 13.5311(7), c = 16.0711(8)
Å, β = 110.439(1)°, V = 3377.3(3)
Å3, Z = 4, 8154 independent reflections,
θmax = 28.3°, 597 parameters, R1 =
0.042, wR2 = 0.112. 3:
C55H61N9O23Cl6Cu
2, M = 1555.9, monoclinic,
P21/n, a = 12.1045(2), b =
33.7116(5), c = 15.8529(2) Å, β =
103.798(1)°, V = 6282.3(2) Å3, Z =
4, 7686 independent reflections, θmax = 22°,
901 parameters, R1 = 0.054, wR2 = 0.154. 4:
C75H75N15.50O14Cl8C
u2, M = 1828.2, triclinic, P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ,
a = 19.3832(5), b = 19.5174(5), c = 24.2065(7)
Å, α = 71.184(2), β = 80.390(2),
γ = 76.041(2)°, V =
8372.5(4)Å3, Z = 4, 28535 independent
reflections, θmax = 24.7°, 2017 parameters,
R1 = 0.069, wR2 = 0.205. CCDC 182/1863. See
http://www.rsc.org/suppdata/cc/b0/b008714i/ for crystallographic
files in .cif format..
,
a = 19.3832(5), b = 19.5174(5), c = 24.2065(7)
Å, α = 71.184(2), β = 80.390(2),
γ = 76.041(2)°, V =
8372.5(4)Å3, Z = 4, 28535 independent
reflections, θmax = 24.7°, 2017 parameters,
R1 = 0.069, wR2 = 0.205. CCDC 182/1863. See
http://www.rsc.org/suppdata/cc/b0/b008714i/ for crystallographic
files in .cif format.. - We were not able to isolate semiquinone species; when using a CuI∶Q stoichiometry of 1∶1, quantitative formation of CuII–chloro compounds and a coupling product between catechol (formed by reduction of the quinone by the CuI complex) and quinone indicated nucleophilic substitution at the catechol; R. M. Buchanan, B. J. Fitzgerald and C. G. Pierpont, Inorg. Chem., 1979, 18, 3439 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: S1–S3: titration data; S4: experimental; S5: colour version of Fig. 1. See http://www.rsc.org/suppdata/cc/b0/b008714i/ |
| This journal is © The Royal Society of Chemistry 2001 |