Solvatochromic response imposed by environmental changes in matrix/chromophore entities: luminescent cyclometalated platinum(II) complex in Nafion and silica materials†
Chi-Ming
Che
*,
Wen-Fu
Fu
,
Siu-Wai
Lai
,
Yuan-Jun
Hou
and
Yun-Ling
Liu
Department of Chemistry and HKU-CAS Joint Laboratory on New Materials, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, China. E-mail: cmche@hku.hk; Fax: +852 2857 1586; Tel: +852 2859 2154
First published on 2nd December 2002
Abstract
Luminescent cyclometalated complex [Pt(L)py]+ (1) immobilised in Nafion film exhibits a solvatochromic shift in emission maximum from 530 to 650 nm upon immersion in ethanol but no effect is detected with aprotic organic solvents, whereas the emission of the [Pt(L)]+ luminophore anchored in silica materials shows a blue shift from ∼665 to 550 nm upon exposure to pentane vapour but no shift is observed for ethanol vapour.
The susceptibility of metal–metal-to-ligand charge transfer (MMLCT) [dσ* → pσ] absorption and emission energies in weakly interacting platinum(II) diimine systems to be affected by subtle changes in the micro-environment has received tremendous attention over the past several years.1 Perturbation of metal–metal and/or ligand–ligand interactions causes changes in photoluminescence excited states from triplet MMLCT (usually at λ > 650 nm) to MLCT (λ 500–550 nm) or vice versa, and this can be a useful operating principle for the design of luminescent sensory devices.2 If the platinum(II) luminophore is embedded inside a host or supporting matrix,3 we envision that host–guest (analyte, such as volatile organic compounds (VOCs)) contacts could affect the metal–metal and/or ligand–ligand interactions and hence the interchange between 3MMLCT and 3MLCT states. Herein is described the employment of the cyclometalated [Pt(L)]+ (HL = 4,6-diphenyl-2,2′-bipyridine) moiety as a luminescent probe in Nafion 417 film and silica materials. Importantly, we demonstrate that the influence of guest molecules upon the nature of the emissive state and energy (3MLCT or/and 3MMLCT) can differ significantly depending on the supporting host material.
The complex [Pt(L)py]+ (1) was prepared according to the literature method.4 Incorporation of 1 into Nafion film was achieved by adopting the previously reported procedures.5 Immobilisation of [Pt(L)]+ moieties into MCM-41/-48 and silica gel were obtained by reacting [Pt(L)Cl] with the modified silica surfaces as depicted in Scheme 1.
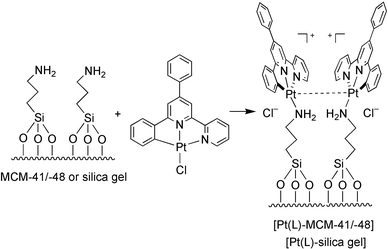 | ||
| Scheme 1 | ||
A series of absorption spectra for the incorporation of 1 into a Nafion film (0.5 × 0.5 cm2) were obtained by the immersion of the Nafion film in an acetonitrile solution of 1 (3 × 10−5 mol dm−3) for extended periods (Fig. 1). The absorbance increases as the loading of 1 proceeds. The inset of Fig. 1 shows the absorbance at 350 nm as a function of loading time, which reaches a plateau after ca. 600 min of loading. The difference between the concentration of the solution before and after loading revealed that ∼0.07 mg of 1 had been immobilised into the Nafion film.
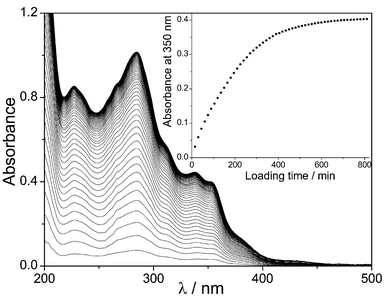 | ||
| Fig. 1 UV–vis absorption spectra of 1 in Nafion at various times (15–810 min) after immersion into an acetonitrile solution of 1 (3 × 10−5 mol dm−3). The spectra represent the difference between the spectra recorded immediately after introducing the film into the solution and those recorded at later times. Inset: absorbance at 350 nm as a function of loading times. | ||
The emission of the modified Nafion film depends on the loading time (see ESI†). After immersing a 0.5 × 0.5 cm2 piece of Nafion film in an acetonitrile solution of 1 (5 × 10−5 mol dm−3) for up to 90 min, the film exhibits an intense emission at λmax 530 nm, which is comparable to the 3MLCT emission of 1 in acetonitrile (λmax 542 nm) at room temperature. As the loading time is increased (120–300 min), the intensity of the 3MMLCT emission at λ > 600 nm increases at the expense of the 3MLCT band at λmax 530 nm. At loading times of more than 300 min, only the 665 nm emission is observed.
We investigated the solvatochromic response of the Nafion film that is not ‘saturated’ with 1 (loading time ∼90 min), for which the intrinsic emission band appears at 530 nm. Upon immersing the film in solvents such as pentane, benzene, acetonitrile or dichloromethane, this emission showed no notable shift in energy (λmax ∼532 nm), whereas in ethanol, the emission maximum significantly red-shifted to 650 nm (Fig. 2). This shift in emission energy in ethanol was reversible and the yellow emission was restored after the film was dried in air for 24 h at room temperature. Such interchange between the 3MLCT (λmax 530 nm) and 3MMLCT (λmax 650 nm) emissions implies that intrusion of ethanol into the Nafion host can significantly alter the metal–metal and/or ligand–ligand interactions between [Pt(L)py]+ units when the Nafion film is partially loaded.‡
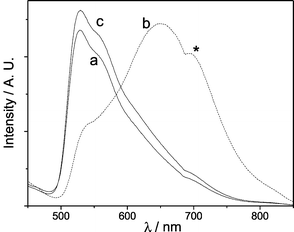 | ||
| Fig. 2 Emission spectra (λex 350 nm) of Nafion film incorporated with 1 (for 90 min) before (a), and after (b) immersion in ethanol for 20 min, and (c) after drying in air for 24 h; * denotes instrumental artifact. | ||
The solvatochromic behaviour of 1 in Nafion film was compared with that of [Pt(L)]+ attached to silica materials.§ The FT Raman spectrum of [Pt(L)Cl] shows the ν(Pt–Cl) stretching at 331 cm−1, which disappears upon immobilisation into MCM-41 and -48. The X-ray powder diffraction patterns of MCM-41, APTES-modified MCM-41, and [Pt(L)-MCM-41] are almost identical, indicating that the structure of MCM-41 is retained.6 The platinum content ([Pt]) in [Pt(L)-MCM-41] determined by ICP atomic emission spectroscopy lies in the range 0.18–5.36 wt%, and increases with the concentration of [Pt(L)Cl] used in the incorporation procedure. When platinum loading is low ([Pt] < 0.4 wt%), excitation at 380 nm results in an emission band at λmax 550 nm (τ = 1.6 μs) which is comparable to the 3MLCT emission (λmax 550 nm, τ = 1.0 μs) of [Pt(L)NH2tBu]ClO4 in acetonitrile. As [Pt] is increased, an emission band at λmax > 660 nm increases in intensity at the expense of the 550 nm emission. At [Pt] > 2.8 wt%, only the emission at 670 nm (τ1 = 0.24 μs, τ2 = 0.88 μs) is observed and this is assigned to a 3MMLCT excited state. Similar findings were noted using MCM-48 instead of MCM-41 (see ESI†).
Photoluminescence responses of [Pt(L)-MCM-41] and [Pt(L)-MCM-48] towards solvents were investigated using [Pt] of 2.8 wt%, which displays a red emission at λmax 662 nm at 298 K. The [Pt(L)-MCM-41/-48] materials were placed in an air-tight tank saturated with vapours of a low-polarity solvent such as pentane, benzene, chloroform, or dichloromethane for 10 min, and a substantial blue-shift of the emission maximum to 548 nm was observed (Fig. 3). This process was reversible and the red emission was restored to its original intensity after the sample was removed from the tank and dried in air for 3 d. In contrast to the Nafion material, there was no noticeable shift in emission energy when [Pt(L)-MCM-41/-48] was similarly exposed to ethanol or methanol vapour. Similar vapochromic behaviour towards low-polarity solvents was observed for [Pt(L)-silica gel] with [Pt] of 2.5 wt% (see ESI†), but in this case, incomplete conversion of the 3MMLCT to 3MLCT emission was observed. We expect that the high surface area and large pore volume of the MCM-41/-48 supports3 will facilitate rapid and reversible sorption/desorption of solvent molecules into the channels of the [Pt(L)-MCM-41/-48] materials, and we ascribe the lower optical response of [Pt(L)-silica gel] towards low-polarity solvents to the amorphous nature of the surface structure.
![Emission spectra (λex 380 nm) of [Pt(L)-MCM-41] ([Pt] = 2.8 wt%) before (a), and after (b) exposure to pentane vapour at 298 K.](/image/article/2003/CC/b210284f/b210284f-f3.gif) | ||
| Fig. 3 Emission spectra (λex 380 nm) of [Pt(L)-MCM-41] ([Pt] = 2.8 wt%) before (a), and after (b) exposure to pentane vapour at 298 K. | ||
The dissimilar solvato-/vapochromic behaviour of the [Pt(L)]+ sensory probe when incorporated into different supports deserves further discussion. The [Pt(L)]+ chromophore in Nafion film revealed a solvatochromic shift towards ethanol only, while embedding this luminescent probe into silica materials resulted in sensitivity towards low-polarity solvents but not alcohols. This demonstrates that the sensing mechanism of the emissive component [Pt(L)]+ is different when the supporting matrix is changed. Possible interaction of ethanol with the sulfonate ionic sites or fluorocarbon phase of the Nafion membrane7 may affect the remote phenyl rings attached to the cyclometalated C⁁N⁁N ligands leading to aggregation of the luminophore 1, and hence the low-energy 3MMLCT emission. On the contrary, interaction between silica materials and alcohols could be less prevalent. Instead, the high sorption capacity of silica materials for low-polarity or nonpolar solvents such as benzene8 and pentane could lead to disruption and segregation of the metal–metal and/or ligand–ligand interactions among the incorporated [Pt(L)]+ moieties. It is apparent that the resulting matrix/solvent interaction consequently influences the micro-environment of the Pt(II) luminophore, and hence the metal–metal and/or ligand–ligand interactions, leading to a switching phenomenon. Thus, anchoring luminescent platinum(II) probes onto a variety of functionalised supporting frameworks could pave the way for new developments in luminescent sensory materials.
We are grateful for financial support from The University of Hong Kong, the Croucher Foundation of Hong Kong, the HKU Large Item Equipment Grant and the Research Grants Council of the Hong Kong SAR, China [HKU 7298/99P].
Notes and references
- W. Paw, S. D. Cummings, M. A. Mansour, W. B. Connick, D. K. Geiger and R. Eisenberg, Coord. Chem. Rev., 1998, 171, 125 CrossRef CAS; M. Kato, C. Kosuge, K. Morii, J. S. Ahn, H. Kitagawa, T. Mitani, M. Matsushita, T. Kato, S. Yano and M. Kimura, Inorg. Chem., 1999, 38, 1638 CrossRef CAS.
- H. Q. Liu, T. C. Cheung and C. M. Che, Chem. Commun., 1996, 1039 RSC; L. Z. Wu, T. C. Cheung, C. M. Che, K. K. Cheung and M. H. W. Lam, Chem. Commun., 1998, 1127 RSC.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56 CrossRef CAS.
- S. W. Lai, M. C. W. Chan, T. C. Cheung, S. M. Peng and C. M. Che, Inorg. Chem., 1999, 38, 4046 CrossRef CAS.
- E. Sabatani, H. D. Nikol, H. B. Gray and F. C. Anson, J. Am. Chem. Soc., 1996, 118, 1158 CrossRef CAS.
- U. Junges, W. Jacobs, I. Voigt-Martin, B. Krutzsch and F. Schüth, J. Chem. Soc., Chem. Commun., 1995, 2283 RSC.
- Perfluorinated Ionomer Membranes, ed. A. Eisenberg and H. L. Yeager, ACS Symposium Series No. 180, American Chemical Society, Washington, DC, 1982 Search PubMed.
- J. C. Vartuli, A. Malek, W. J. Roth, C. T. Kresge and S. B. McCullen, Microporous Mesoporous Mater., 2001, 44–45, 691 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- C. J. Liu, W. Y. Yu, S. G. Li and C. M. Che, J. Org. Chem., 1998, 63, 7364 CrossRef CAS.
- U. Schubert, New J. Chem., 1994, 18, 1049 Search PubMed.
- Y. G. Ma, T. C. Cheung, C. M. Che and J. C. Shen, Thin Solid Films, 1998, 333, 224 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: characterisation data for 1 and [Pt(L)NH2tBu]ClO4, and emission spectra for various loadings of [Pt(L)]+ on different supports and solvato-/vapochromic responses. See http://www.rsc.org/suppdata/cc/b2/b210284f/ |
| ‡ Photoluminescence response towards solvents was also examined using Nafion films with high loading of 1 (loading time > 400 min). The emission at 653 nm displayed no notable shift in energy upon immersing the film in ethanol. This suggests that when a Nafion film is fully loaded with 1, the effect upon the intermolecular metal–metal and/or ligand–ligand interactions is less apparent. |
| § MCM-41 and -48 were prepared by literature methods.9 The modified MCM-41/-48 with (3-aminopropyl)triethoxysilane (APTES)10 (100 mg) was stirred with [Pt(L)Cl] (0.7–25 mg) in CHCl3 (10 mL) for 8 h at room temperature. The resultant [Pt(L)-MCM-41/-48] was washed with CHCl3 and dried. Incorporation of [Pt(L)]+ moieties into APTES-modified silica gel11 was similarly obtained.12 |
| This journal is © The Royal Society of Chemistry 2003 |