Mechanistic aspects of acetone addition to metalloaromatic complexes of iridium: a DFT investigation†
Mark A.
Iron
,
Jan M. L.
Martin
* and
Milko E.
van der Boom
*
Department of Organic Chemistry, Weizmann Institute of Science, 76100, Rehovot, Israel. E-mail: comartin@wicc.weizmann.ac.il, milko.vanderboom@weizmann.ac.il; Fax: +972 8 934 4142; Tel: +972 8 934 3829
First published on 3rd December 2002
Abstract
DFT calculations were used to reveal the unexpected reactivity and mechanism of the addition of acetone to metallabenzene, metallapyrylium and metallathiabenzene complexes of iridium.
The discovery of the aromatic nature of benzene was a landmark discovery in chemistry. The unique properties of aromatic compounds have driven many chemists to substitute aromatic CH units. Initial goals included ‘organic’ moieties such as O, N, P and S. Later targets involved using main group metals (e.g., B,1a Si,1b Ge,1c As1d,e) and even transition metals (e.g., Os,2a,b Ru,2c–e Pt,2f Ni,2g Mo,2h Ir2i,j,3,4). There is also a metallabenzyne complex of osmium.5 Despite the significant amount of experimental data available on metalloaromatic complexes,2–5 theoretical6 and kinetic studies are scarce.4
Recently, Bleeke et al. published an interesting series of papers on the synthesis and unusual reactivity of metalloaromatic complexes of Ir(I), [Me2C4H2XIr(PEt3)3]n+, specifically a metallabenzene (1C, X = CH, n = 0),3b a metallapyrylium (1O, X = O, n = 1, as BF4− salt)3c,d and a metallathiabenzene (1S, X = S, n = 1, as BF4− salt).3e,f The fascinating reactions of these complexes with a variety of organic substrates is intriguing yet not well understood. For instance, the metallabenzene (1C) and metallathiabenzene (1S) complexes are unreactive towards acetone while the metallapyrylium complex (1O) reversibly adds acetone in a 1,4-fashion [eqn. (1)]. The facile reversibility of this reaction and the colour change observed upon dissolving the purple metallapyrylium complex in acetone to give the yellow adduct (2O) may be useful in several applications, including chemical sensors.
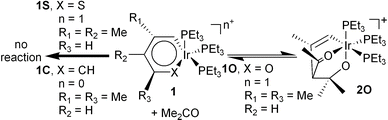 | (1) |
We report herein the 1,4-addition of acetone to the model complexes [C4H4XIr(PH3)3]n+ (3C, X = CH, n = 0; 3O, X = O, n = 1; 3S, X = S, n = 1) using DFT methods (Fig. 1). We selected the mPW1K7/SDB-cc-pVDZ//mPW1K/SDD level of theory having previously recommended its use in investigating reaction mechanisms.8 All calculations used Gaussian 989 (see ESI† for full computational details). The identities of the transition states were confirmed by intrinsic reaction coordinate (IRC) calculations.
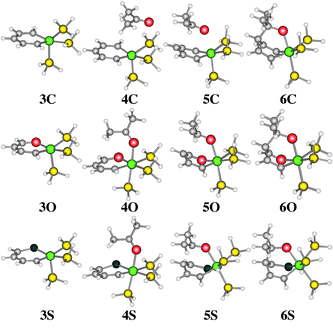 | ||
| Fig. 1 Calculated structures of complexes 3–6. Colour scheme: Ir: green, C: grey, H: white, O: red. S: blue, P: yellow. | ||
A stepwise and a concerted reaction mechanism for the reversible acetone addition to 3 are considered here. In the former, an intermediate would be formed prior to C–C bond formation where the acetone is η1-coordinated to the metal. In the latter, the Ir–O and C–C bonds are formed in a concerted (Diels–Alder) fashion. At present, it is unknown which mechanism transpires.
The metalloaromatic complexes 3 were optimised and they have a square pyramidal geometry with apical phosphine ligands. The calculated C–C bond lengths show little variation and are all near the calculated value for benzene (1.396 Å). In fact, the Cortho–Cmeta and Cmeta–Cpara lengths in 3C are 1.395 and 1.398 Å, respectively. The calculated structures of 3C and 3S agree satisfactorily with the reported X-ray structures of 1C3b and 1S.3e In all three cases, the six-membered rings are basically flat as appropriate for aromatic systems.
From 3O and 3S, an η1-coordinated acetone complex is initially formed yielding 4O and 4S, respectively. In both complexes there is some alternation of the ring C–C bonds, suggesting partial loss of aromaticity. In these two complexes, the Ir–O (acetone) bond lengths are 2.131 Å (4O) and 2.140 Å (4S).10 This bond length in 4O gives an indication of the nature of the aromatic Ir–O bond of 3O and 4O. In these two complexes, the ring Ir–O bonds are 1.997 and 2.030 Å, respectively, further emphasizing the aromatic nature of the former and the reduced aromaticity of the latter. In complexes 4O and 4S, the acetone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds have been lengthened from 1.235 Å (free acetone) to 1.256 Å.10 Such an intermediate would be expected for a step-wise addition reaction mechanism.
O bonds have been lengthened from 1.235 Å (free acetone) to 1.256 Å.10 Such an intermediate would be expected for a step-wise addition reaction mechanism.
For 3C, acetone coordination could not be found. Instead, a long-range complex (4C) was located. In this complex, the acetone is not coordinated to the metal centre but rather lies parallel to the metallabenzene plane and to the Ir⋯Cpara axis. The metallabenzene moiety and the acetone CO bond lengths remain unchanged, the latter 1.241 Å. The distances between the two fragments are 2.991 Å (Ir⋯O) and 5.250 Å (C⋯C).
The expected products (6) were identified and exhibit a puckered ring. The alternating ring C–C bond lengths clearly resemble a 2,4-cyclohexadiene system with, in 6C for example, Cortho–Cmeta of 1.340 Å and Cmeta–Cpara of 1.515 Å.
The transition states (5) for the addition of acetone were found and the bond lengths in the transition states are intermediate between the reactants (4) and the products (6). For 4O and 4S, the acetone ligand has rotated so that the methyl groups are parallel to the metalloaromatic ring and the acetone ligand is now bent towards the ring. The Cpara–Ir–O angle decreases from 100.0 to 60.6 to 53.4° for X = O and 102.4 to 55.8 to 51.3° for X = S. Furthermore, the ring becomes puckered during the transition state (5), but to a lesser degree than in the product complexes (6). The two transition states are characterised by a single imaginary vibrational frequency whose normal coordinate corresponds to C–C bond formation. Overall, the obtained transition states (4O and 4S) are what would be expected for stepwise addition of acetone across the metalloaromatic system.
For the X = CH system, the transition state (5C) was also as expected. Again, the bond lengths are intermediate between 4C and 6C. The acetone moiety remains basically parallel to the metallabenzene plane throughout the reaction. The metallabenzene ring has become puckered and the Ir⋯O and C⋯C distances have decreased to 2.411 and 2.689 Å, respectively. This transition state, characterised by a single imaginary frequency for simultaneous Ir–O and C–C bond formations, is indicative of a concerted reaction mechanism.
The calculated structures of the complexes 3–6 are depicted in Fig. 1. One can clearly understand the observed experimental results [eqn. (1)]3 from the energetic data for the three reactions in Table 1. Of the three complexes (3), only the metallapyrylium (3O) reacts with acetone. This reaction has the lowest reaction barrier with ΔG‡298 = 14.5 kcal mol−1. The fact that the overall reaction is only mildly exothermic, with ΔG298 = −6.8 kcal mol−1, is essential for the process to be reversible. If the reaction would be too exothermic, the barrier for the reverse reaction would be too large. In this case, ΔG‡298 = 13.3 kcal mol−1 for the reverse reaction.
| C | O | S | ||||
|---|---|---|---|---|---|---|
| Complex | ΔE | ΔG298 | ΔE | ΔG298 | ΔE | ΔG298 |
| 3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 4 | −6.7 | 3.3 | −22.5 | −7.9 | −21.9 | −7.6 |
| 5 | 18.6 | 33.2 | −9.5 | 6.6 | −0.2 | 15.8 |
| 6 | −17.8 | −0.2 | −22.4 | −6.8 | −9.8 | 6.7 |
On the other hand, the situation is not as favourable for the other two systems. For the metallathiabenzene (3S), the reaction barrier is not unreasonably high at room temperature. Nonetheless, the overall reaction is endothermic and, therefore, the rate of the reverse reaction would be approximately 14 orders of magnitude faster. In the case of the metallabenzene (1C), the reaction barrier is simply too high to allow the reaction to proceed, even though the overall reaction is thermoneutral.
The main question that remains is why there is this remarkable difference in reactivities between 3 and acetone. There are two potential contributions to the observed reaction pattern. The metallabenzene complex (3C) is neutral while the other two (3O and 3S) are cationic. A significant stabilisation can be obtained by coordination of the electronegative oxygen of acetone to the cationic metal centre. In fact, in all three cases the formation of 4 is exoenergetic, but the formation of 4C is not sufficiently so to overcome the loss of entropy and the formation of 4C is thus slightly endothermic. The formation of the η1-coordinated acetone complexes 4O and 4S directs, and thus facilitates, the addition reaction.
The second important factor is the Ir⋯Cpara diagonal of the metalloaromatic ring. The acetone CO bond has to stretch across the ring during the reaction, and this is energetically unfavourable. In the transition state, the CO bond of acetone are 1.277 Å (5C), 1.313 Å (5O) and 1.325 Å (5S). The Ir⋯Cpara distances for 3O, 3C and 3S are 3.400, 3.456 and 3.602 Å, respectively. Not surprisingly, this follows the same order as the single bond covalent radii of 0.66, 0.77 and 1.05 Å for O, C and S.11
In summary, the addition of acetone was investigated for a series of isostructural metalloaromatic complexes of Ir(I). The calculated reactivities fully agree with the experimental observations.3 It was found that 1O and 1S follow a step-wise mechanism while 1C would react in a concerted manner. The addition of acetone to the metallathiabenzene (3S) does not proceed because of the high barrier caused by the relatively large ring size and the endothermicity of the reaction. Both the larger ring size of the metallabenzene complex (3C) and the non-concerted transition state make the addition to this metal centre difficult. We are currently examining other addition reactions to this class of complexes.
Research was supported by the Helen and Martin Kimmel Center for Molecular Design, by the Tashtiyot (Infrastructures) program of the Ministry of Science and Technology (Israel) and by a computer time grant from the (Israel) Inter-University Computer Center. M. A. I. acknowledges a Doctoral Fellowship from the Feinberg Graduate School (Weizmann Institute of Science).
Notes and references
- (a) G. E. Herberich, U. Englert and D. Pubanz, J. Organomet. Chem., 1993, 459, 1 CrossRef CAS; (b) K. Wakita, N. Tokitoh, R. Okazaki, S. Nagase, P. von Ragué Schleyer and H. Jiao, J. Am. Chem. Soc., 1999, 121, 11336 CrossRef CAS; (c) N. Nakata, N. Takeda and N. Tokitoh, J. Am. Chem. Soc., 2002, 124, 6914 CrossRef CAS; (d) A. J. Ashe III, Top. Curr. Chem., 1982, 105, 125 Search PubMed; (e) C. Elschenbroich, J. Kroker, W. Massa, M. Wünsch and A. J. Ashe III, Angew. Chem., Int. Ed. Engl., 1986, 25, 571 CrossRef.
- (a) G. P. Elliott, W. R. Roper and J. M. Waters, J. Chem. Soc., Chem. Commun., 1982, 811 RSC; (b) C. E. F. Rickard, W. R. Roper, S. D. Woodgate and L. J. Wright, Angew. Chem., Int. Ed. Engl., 2000, 39, 750 CrossRef CAS; (c) W. Lin, S. R. Wilson and G. S. Girolami, Organometallics, 1997, 16, 2356 CrossRef CAS; (d) S. H. Liu, W. S. Ng, H. S. Chu, T. B. Wen, H. Xia, Z. Y. Zhou, C. P. Lau and G. Jia, Angew. Chem., Int. Ed., 2002, 41, 1589 CrossRef CAS; (e) U. Englert, F. Podewils, I. Schiffers and A. Salzer, Angew. Chem., Int. Ed., 1998, 37, 2134 CrossRef CAS; (f) V. Jacob, T. J. R. Weakley and M. M. Haley, Angew. Chem., Int. Ed., 2002, 41, 3470 CrossRef CAS; (g) U. Bertling, U. Englert and A. Salzer, Angew. Chem., Int. Ed. Engl., 1994, 33, 1003 CrossRef; (h) M. S. Kralik, A. L Rheingold, J. P. Hutchinson, J. W. Freeman and R. D. Ernst, Organometallics, 1996, 15, 551 CrossRef CAS; (i) R. D. Gilbertson, T. J. R. Weakley and M. M. Haley, J. Am. Chem. Soc., 1999, 121, 2597 CrossRef CAS; (j) R. D. Gilbertson, T. J. R. Weakley and M. M. Haley, Chem. Eur. J., 2000, 6, 437 CrossRef CAS.
- (a) J. R. Bleeke, Y.-F. Xie, W.-J. Peng and M. Chiang, J. Am. Chem. Soc., 1989, 111, 4118 CrossRef CAS; (b) J. R. Bleeke, R. Behm, Y. –F. Xie, M. Y. Chiang, K. D. Robinson and A. M. Beatty, Organometallics, 1997, 16, 606 CrossRef CAS; (c) J. R. Bleeke and J. M. B. Blanchard, J. Am. Chem. Soc., 1997, 119, 5443 CrossRef CAS; (d) J. R. Bleeke, J. M. B. Blanchard and E. Donnay, Organometallics, 2001, 20, 324 CrossRef CAS; (e) J. R. Bleeke and P. V. Hinkle, J. Am. Chem. Soc., 1999, 121, 595 CrossRef CAS; (f) J. R. Bleeke and P. V. Hinkle, Organometallics, 2001, 20, 1939 CrossRef CAS.
- For reviews, see: (a) J. R. Bleeke, Chem. Rev., 2001, 101, 1205 CrossRef CAS; (b) J. R. Bleeke, Acc. Chem. Res., 1991, 24, 271 CrossRef CAS.
- (a) T. B. Wen, Z. Y. Zhou and G. Jia, Angew. Chem., Int. Ed., 2001, 40, 1951 CrossRef CAS; (b) W. R. Roper, Angew. Chem., Int. Ed., 2001, 40, 2440 CrossRef CAS.
- (a) D. L. Thorn and R. Hoffmann, Nouv. J. Chim., 1979, 3, 39 Search PubMed; (b) H. Masui, Coord. Chem. Rev., 2001, 219–221, 957 CrossRef CAS; (c) K. K. Baldridge, O. Uzan and J. M. L. Martin, Organometallics, 2000, 19, 1477 CrossRef CAS.
- J. B. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811 CrossRef CAS.
- M. A. Iron, H. C. Lo, J. M. L. Martin and E. Keinan, J. Am. Chem. Soc., 2002, 124, 7041 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 98, Revision A.11, Gaussian Inc., Pittsburgh PA, 2001.
- These values agree with X-ray characterised Ir–acetone complexes. For example, see: (a) M. V. Jiménez, E. Sola, J. A. López, F. J. Lahoz and L. A. Oro, Chem. Eur. J., 1998, 4, 1398 CrossRef CAS; (b) D.-H. Lee, J. Chen, J. W. Faller and R. H. Crabtree, Chem. Commun., 2001, 213 RSC.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem Soc., Perkin Trans. 2, 1987, S1 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: selected geometric data, calculated structures of all complexes and full computational details. See http://www.rsc.org/suppdata/cc/b2/b210622a/. |
| This journal is © The Royal Society of Chemistry 2003 |