A new route to furanoeremophilane sesquiterpenoids. Synthesis of (±)-6β-hydroxyeuryopsin
M. Sundaram
Shanmugham
and
James D.
White
*
Department of Chemistry, Oregon State University, Corvallis, Oregon 97331-4003, USA. E-mail: james.white@orst.edu; Fax: 541-737-2660; Tel: 541-737-2173
First published on 21st November 2003
Abstract
The naturally occurring furanoeremophilane 6β-hydroxyeuryopsin was synthesized by a novel route which involved Stille coupling of a 2-furylstannane with a cyclohexylmethyl bromide, followed by intramolecular formylation of the furan to complete the tricyclic nucleus of the sesquiterpenoid.
The eremophilane family is a large, structurally diverse group of sesquiterpenoids characterized by a decalin skeleton in which a methyl migration has taken place to produce a non-isoprenoid substituent pattern.1 A subset of this group, the furanoeremophilanes, bears a furan fused to the decalin core which, in certain cases, appears in oxidized form as a butenolide.
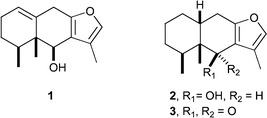
Beginning in 1971 with Piers' pioneering synthesis of (±)-eremophilenolide,2 a substantial synthetic effort has been devoted to this class of natural products with the result that several distinct pathways have been established.3 However, none of these builds the furanoeremophilane framework through consecutive alkylations at C2 and then C3 of a preformed furan. We now describe a synthesis of 6β-hydroxyeuryopsin (1), a furanoeremophilane isolated from Senecio tolucannus,4 which exemplifies just such a strategy, and which, in principle, allows access to a broad array of related sesquiterpenoids such as petasalbine (2) and ligularone (3).5
The synthesis of 1 commenced from the known 2,3-dimethyl-2-methallylcyclohexanone (4),6 prepared as a 4 : 1 mixture of cis and trans isomers from 2,3-dimethylcyclohexanone (Scheme 1). Ketalization of the major isomer 4 was selective and was followed by acid-catalyzed isomerization of the terminal alkene to yield the trisubstituted olefin 5. This was subjected to oxidative cleavage to give aldehyde 6.7 The latter was reduced, and the resulting primary alcohol 7 was protected as its triisopropylsilyl (TIPS) ether 8. Mild acidic hydrolysis of ketal 8 under conditions that left the TIPS ether intact produced a ketone which was condensed with 2,4,6-triisopropylbenzenesulfonylhydrazine. Shapiro reaction8 of hydrazide 9 with tert-butyllithium, followed by treatment of the intermediate lithio alkene with dimethylformamide, afforded α,β-unsaturated aldehyde 10 which was reduced to primary alcohol 11. This compound was converted via its mesylate 12 to allylic bromide 13.
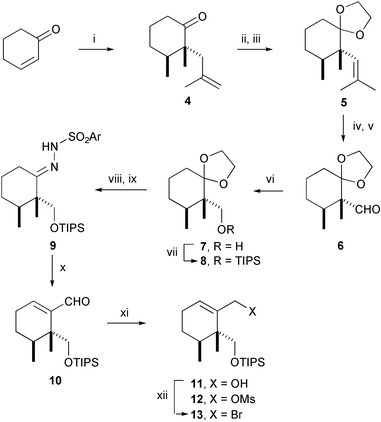 | ||
| Scheme 1 Reagents and conditions: i, Ref. 6; ii, 2-ethyl-2-methyl-1,3-dioxolane, (CH2OH)2, p-TsOH (0.2 equiv.), rt, 72 h, 75%; iii, p-TsOH (0.05 equiv.), benzene, 50 °C, 12 h, 57%; iv, K2OsO4 (0.05 equiv.), K2Fe(CN)4, K2CO3, quinolidine, MeSO2NH2, t-BuOH–H2O, rt, 48 h, 90%; v, NaIO4, THF, H2O, rt, 12 h, 100%; vi, NaBH4, THF–H2O, rt, 12 h; vii, TIPSOTf, 2,6-lutidine, CH2Cl2, −78 °C to 0 °C, 4 h, 93%; viii, PPTS (0.3 equiv.), 10% aq. acetone, 60 °C, 4 h, 85%; ix, 2,4,6-triisopropylbenzenesulfonylhydrazine, THF, rt, 12 h, 100%; x, tert-BuLi, 10% TMEDA–hexanes, −78 °C, 30 min, then 0 °C, 1 min, then −78 °C, DMF, −78 °C to 0 °C, 4 h; xi, DIBALH, CH2Cl2, −78 °C to 0 °C, 4 h, 75% from 9; xii, Ms2O, Et3N, CH2Cl2, −78 °C to 0 °C, 2 h, then LiBr, THF, rt, 12 h, 95%. | ||
The furanoid partner required for coupling with 13 was obtained from furan-3-carboxylic acid (14) (Scheme 2). After reduction of this acid with borane and conversion of the resulting primary alcohol to tert-butyldimethylsilyl (TBS) ether 15, the furan was reacted with n-butyllithium in HMPA to yield 16. This transformation is presumed to occur via an intramolecular retro-Brook rearrangement9 of the corresponding 2-lithiofuran and conveniently blocks C-2 of the furan against further substitution at this position. After reductive cleavage of the mesylate of 16 with lithium triethylborohydride, lithiation of the furan with n-butyllithium took place exclusively at C-5, and subsequent addition of tri-n-butyltin chloride10 cleanly furnished the furyl stannane 17.
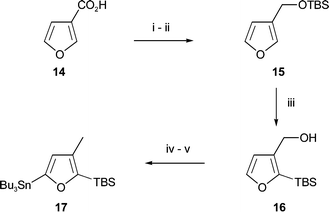 | ||
| Scheme 2 Reagents and conditions: i, BH3·Me2S, THF, rt, 12 h, 85%; ii, TBSCl, imidazole, CH2Cl2, rt, 2 h, 100%; iii, n-BuLi, HMPA–THF, −78 °C to rt, 6 h, 89%; iv, Ms2O, Et3N, CH2Cl2, −78 °C to 0 °C, 2 h, then LiEt3BH, THF, 0 °C, 100%; v, n-BuLi, THF, −78 °C to 0 °C, 6 h, then n-Bu3SnCl, −78 °C to rt, 12 h, 85%. | ||
Stille coupling11 of allylic bromide 13 with stannane 17 was carried out in the presence of catalytic palladium(0) dibenzylideneacetone complex and triphenylarsine to furnish the alkylfuran 18 in good yield (Scheme 3). Removal of the TIPS ether and oxidation of the resulting primary alcohol 19 to aldehyde 20 set the stage for closure to the tricyclic eremophilane skeleton. After considerable experimentation with a variety of Lewis acids, it was found that cyclization of 20 could be accomplished in quantitative yield with trimethylsilyl triflate in the presence of 2,6-lutidine. The initial product was the trimethylsilyl (TMS) ether 21 accompanied by 15% of its 6α epimer. Selective cleavage of the TMS ether from this mixture produced alcohol 22 which formed a crystalline p-nitrobenzoate ester, and X-ray crystallographic analysis of this derivative fully confirmed the stereostructure of 22 (Fig. 1).† Removal of the TBS ether from the furan moiety of 22 required strenuous conditions but was accomplished with a 2 M solution of TBAF in THF9 and gave a substance identical with the natural product 6β-hydroxyeuryopsin (1) based on comparison of NMR spectra.4
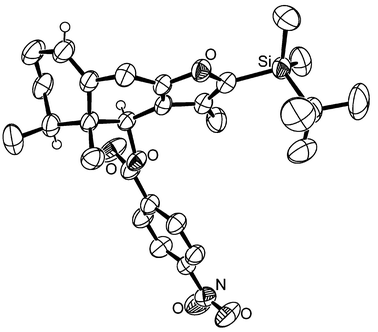 | ||
| Fig. 1 ORTEP plot of the crystal structure of the p-nitrobenzoate of 22. Thermal ellipsoids are drawn at the 30% probability level. | ||
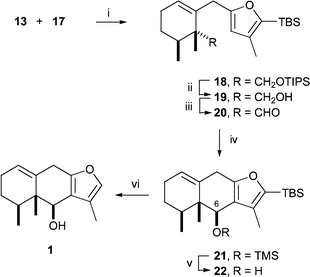 | ||
| Scheme 3 Reagents and conditions: i, Pd2(dba)3 (0.2 equiv.), AsPh3 (0.8 equiv.), THF, rt, 48 h; ii, TBAF, THF, rt, 12 h, 85% from 13; iii, TPAP (0.05 equiv.), 4 Å mol sieves, N-methylmorpholine-N-oxide, CH2Cl2, rt, 2 h, 85%; iv, TMSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 12 h, 100%; v, TBAF, THF, rt, 2 h; vi, 2 M TBAF (20 equiv.), THF, rt, 24 h, 60% from 21. | ||
In summary, a new pathway to furanoeremophilanes has been established which constructs the tricyclic framework from a 2-alkylfuran through closure of the central cyclohexane ring. As exemplified in the synthesis of 1, the route exhibits good control of relative stereochemistry and is characterized by a novel tactic for directing substitution at a reactive furan.
We are indebted to Professor Alexandre F. T. Yokochi of this Department for the X-ray crystal structure, and to Professor Romo de Vivar, Universidad Nacional Autónoma de México, for NMR spectra of natural 6β-hydroxyeuryopsin. Financial support was provided by the National Science Foundation (01076103-CHE).
Notes and references
- A. R. Pinder, in Progress in the Chemistry of Organic Natural Products, W. Herz, H. Grisebach, G. W. Kirby, Eds., Springer Verlag: New York, 1977, Vol. 34, p. 81 Search PubMed.
- E. Piers, M. B. Geraghty and R. D. Smillie, J. Chem. Soc., Chem. Commun., 1971, 614 RSC.
- For a review of furanoeremophilane synthesis, see C. H. Heathcock, S. L. Graham, M. C. Pirrung, F. Plavac, C. T. White, in The Total Synthesis of Natural Products, J. ApSimon, Ed., Wiley: New York, 1983, Vol. 5, p. 202 Search PubMed.
- A. L. Arciniegas, G. Pérez-Castorena, J. L. Parada, J. L. Villaseñor and A. Romo De Vivar, Rev. Latinoam. Quim., 2000, 28, 131 Search PubMed.
- H. Ishii, T. Tozyo and H. Minato, Tetrahedron, 1965, 21, 2605 CrossRef CAS.
- E. Piers, R. W. Britton and W. De Waal, Can. J. Chem., 1969, 47, 831 CAS.
- P. Wipf, Y. Kim and D. M. Goldstein, J. Am. Chem. Soc., 1995, 117, 11106 CrossRef CAS.
- A. R. Chamberlin, J. E. Stemke and F. T. Bond, J. Org. Chem., 1978, 43, 147 CrossRef.
- E. Bures, P. G. Spinazzé, G. Beese, I. R. Hunt, C. Rogers and B. A. Keay, J. Org. Chem., 1997, 62, 8741 CrossRef CAS; E. Bures, J. A. Nieman, S. Yu, P. G. Spinazzé, I. R. Hunt, A. Rauk and B. A. Keay, J. Org. Chem., 1997, 62, 8750 CrossRef CAS.
- J. A. Nieman and B. A. Keay, Tetrahedron Lett., 1994, 35, 5335 CrossRef CAS.
- K. Fugami and M. Kosugi, Top. Curr. Chem., 2002, 219, 87 CAS; V. Farina, V. Krishnamurthy and W. Scott, Org. React., 1997, 50, 1 CAS.
Footnote |
† Crystal data for p-nitrobenzoate of22: M
= 495.68; triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; a
= 7.479(2), b
= 8.389(3), c
= 22.545(9)
Å, β
= 99.07(3)°, V
= 1379.1 (8) A3, T
= 293(2) K; Z
= 2; µ
=
(Cu-Kα)
= 1.045 mm−1; reflections: total = 5964, unique 4750 (Rint 0.0279); residuals (all data Shelxl): R1 = 0.0803, wR2 = 0.2000. CCDC 220193. See http://www.rsc.org/suppdata/cc/b3/b311211j/ for crystallographic data in .cif or other electronic format. ; a
= 7.479(2), b
= 8.389(3), c
= 22.545(9)
Å, β
= 99.07(3)°, V
= 1379.1 (8) A3, T
= 293(2) K; Z
= 2; µ
=
(Cu-Kα)
= 1.045 mm−1; reflections: total = 5964, unique 4750 (Rint 0.0279); residuals (all data Shelxl): R1 = 0.0803, wR2 = 0.2000. CCDC 220193. See http://www.rsc.org/suppdata/cc/b3/b311211j/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2004 |