Terminally functionalized polyisobutylene oligomers as soluble supports in catalysis†
David E.
Bergbreiter
* and
Jun
Li
Department of Chemistry, PO Box 30012, Texas A&M University, College Station, Texas, USA. E-mail: bergbreiter@tamu.edu; Fax: 001 979 845 4719; Tel: 001 979 845 3437
First published on 27th November 2003
Abstract
A new phase selective hydrocarbon soluble polymer support is described.
Soluble polymer supports are of increasing interest in synthesis and catalysis.1 Most past attention on polymer supports has focused on polystyrene with catalysts/reagents/ligands on pendant groups.2 Other soluble polar and nonpolar supports which are useful in catalysis and have tunable phase selective solubility that is altered by the identity of alkyl substituent groups have been described.3–8 We and others have shown that such polymers can be recovered by liquid/liquid separations.3–10 Terminally functionalized polymers are soluble alternatives to polymer supports with pendant groups. Poly(alkene oxide) (PEG) is the most common example of such a support.11 These linear polyethers are easier to characterize and can be recovered in the polar phase of a thermomorphic mixture.3 However, PEG supports are more often recovered by less efficient solvent precipitation schemes. Solvent precipitation can be avoided by using terminally functionalized polyethylene (PE) oligomers. PE oligomers are recoverable by precipitation and filtration without excess solvent addition.12 However, PE's insolubility at < 50 °C and solubility only at > 75 °C in nonpolar solvents limit its utility. Here we describe an alternative to either PEG or PE – polyisobutylene (PIB). Suitable terminally functionalized polyisobutylenes are easily prepared from commercially available materials. These readily soluble oligomers and the catalysts attached to them can be easily separated from products by liquid/liquid separation using a nonpolar solvent after a reaction.
The PIB oligomers we used were obtained from BASF as oligomers with a d.p. of ca. 20 or 40 with terminal vinyl groups. As shown in Scheme 1, such groups were converted into ligands for transition metal catalysts or into catalyst precursors using simple transformations. Such ligands have loadings of ca. 1 or 0.5 mmol of ligands/g, loadings that are comparable to polar PEG-based materials. Of particular note is the facility with which these materials can be analyzed by 1H NMR spectroscopy. As shown in Fig. 1, the 1H NMR spectra of these oligomers' end groups are as well resolved as those of a small molecule, simplifying analysis of the course of a reaction.
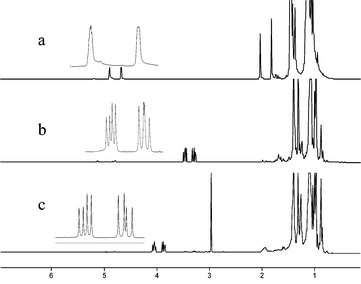 | ||
| Fig. 1 1H NMR spectra of polyisobutylene oligomers: a) 1, with terminal vinyl groups; b) 2, with a terminal –CH2OH group; and c) 3, with a terminal –CH2OSO2CH3 group. The expansions overlaid on these spectra cover 0.4 δ in each case. | ||
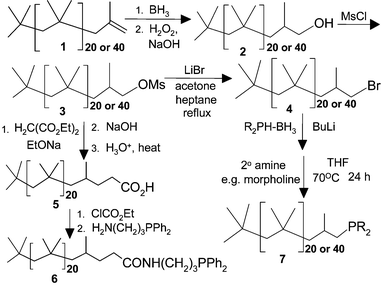 | ||
| Scheme 1 Synthesis routes to terminally functionalized polyisobutylene oligomers containing ligands and catalysts. | ||
To be effective as a support, polyisobutylene (PIB) oligomer separation from the products of a reaction has to be simple and effective. A nominal value for an effective separation is 200 : 1 (< 0.5% loss of catalyst/ligand per stage). Higher separation numbers are more desirable. By preparing PIB oligomers with dansyl or azo dye labels (8 and 9) we have shown that PIB supports are selectively separated into one phase of a thermomorphic biphasic mixture. For example, in 20 mL of a thermomorphic 1 : 1 (v : v) heptane–90% aq. EtOH mixture, 30 mg of 8 was selectively soluble in the heptane-rich phase to an extent of > 99.6%. Fluorescence analysis of a similar solution containing 5 mg of 9 showed that 9 was selectively soluble in the heptane-rich phase to the extent of > 300 : 1.
To demonstrate the utility of PIB as a soluble polymer support, we have looked at several known catalytic reactions. Specifically, we briefly examined three different Pd-catalysts (eqns. (1) and (2)). The first of these was a Pd(II) catalyst or catalyst precursor – the terminally bound pincer sulfur–carbon–sulfur SCS-Pd(II) species 10.13 This Pd catalyst was attached to the PIB oligomer via a terminal –CO2H group. The PIB-bound 10 was then used to carry out Heck chemistry (eqn. (3)). As was true for SCS-Pd(II) species on other polymers, this catalyst was only effective for aryl iodides. However, the catalyst could be used through three cycles without any detectable loss of activity by cooling a reaction mixture to room temperature and separating the heptane rich phase containing the catalyst from the polar phase containing the product. The products were isolated from the polar phase, purified by chromatography and identified by 1H and 13C NMR spectroscopy. Recycling simply involved adding fresh substrate(s) solution to the recovered heptane phase and reheating.
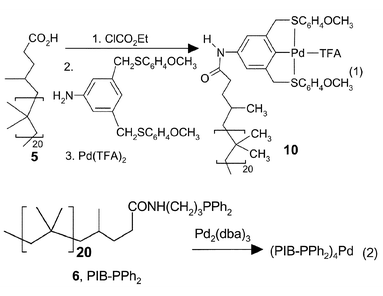
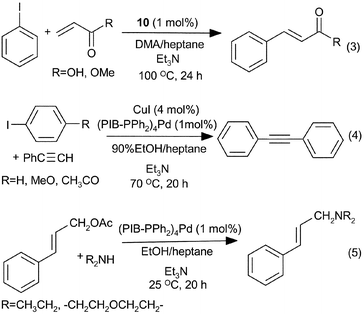
Phosphine-ligated Pd(0) catalysts are generally more reactive than SCS-Pd(II) species.13 Using the reactions shown in eqn. (2), we have attached Pd(0) catalysts to phosphine ligands at the end of a polyisobutylene chain. The resulting phosphine-complexed Pd catalysts were successfully used in two sorts of Pd(0) chemistry – Sonagashira alkyne–arene couplings and allylic substitution (eqns. (4) and (5) respectively). The results of these reactions and catalyst recycling in this chemistry are detailed in Table 1. Recycling followed the general protocol described above. In these cases, recycling required rigorous oxygen free conditions. Otherwise oxidation of the phosphine ligand and formation of Pd black occurred.
| Substrate | Acceptor | Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 |
|---|---|---|---|---|---|
| a Allylic substitutions were carried out in a monophasic EtOH–heptane mixture at 25 °C using 1 mol% (PIB-PPh2)4Pd. Cycle 5 is not shown but recycling was equally effective in cycle 5. Increased yields in cycles 1–3 reflect saturation of the heptane phase by product; Sonogashira reactions were carried out at 70 °C in a monophasic 90% EtOH–heptane mixture using 1 mol% (PIB-PPh2)4Pd; Heck reactions were carried out at 100 °C in a monophasic DMA–heptane mixture using 1 mol% PIB-SCS-Pd (10). These yields are isolated yields | |||||
| cinnamyl acetate | morpholine | 81% | 95% | 99% | 100% |
| cinnamyl acetate | diethylamine | 78% | 99% | 96% | 100% |
| iodobenzene | phenylacetylene | 48% | 95% | 99% | |
| 4-iodoanisole | phenylacetylene | 35% | 97% | 97% | |
| 4′-iodoacetophenone | phenylacetylene | 74% | 95% | 100% | |
| iodobenzene | methyl acrylate | 63% | 98% | 99% | |
| iodobenzene | acrylic acid | 50% | 100% | 100% | |
In summary, the studies here show that polyisobutylene oligomers are a nonpolar, phase selectively soluble alternative to terminally functionalized PEG oligomers for support of catalysts. The derivatization and chemistry of these oligomers can be easily followed by conventional spectroscopy. The activity of the catalysts attached to the oligomers is analogous to that of other soluble polymer or low molecular weight catalysts. Such supports contain only unreactive C–C and C–H bonds (except for the ligating or catalysts species) and should thus be useful as soluble supports for a wide range of catalysts. Finally, these oligomers' nonpolar character ensures that they are readily recoverable in the nonpolar phase by a liquid/liquid separation after either cooling of a thermomorphic solvent mixture or by perturbing a latently biphasic solvent mixture.
Support of this work by the Petroleum Research Fund, the Robert A. Welch Foundation and the National Science Foundation (CHE-0010103) is gratefully acknowledged as is a gift of the PIB oligomers from Dr Rocco Paciello of BASF.
Notes and references
- D. E. Bergbreiter, Chem. Rev., 2002, 102, 3345–3384 CrossRef CAS.
- M. Benaglia, A. Puglisi and F. Cozzi, Chem. Rev., 2003, 103, 3401–3430 CrossRef CAS; C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275–3300 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn, A. Wilson and E. M. Sink, J. Am. Chem. Soc., 2000, 122, 9058–9064 CrossRef CAS; D. E. Bergbreiter, Y.-S. Liu and P. L. Osburn, J. Am. Chem. Soc., 1998, 120, 4250–4251 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn and J. D. Frels, J. Am. Chem. Soc., 2001, 123, 11105–11106 CrossRef CAS.
- D. E. Bergbreiter and C. Li, Org. Lett., 2003, 5, 2445–2447 CrossRef CAS.
- D. E. Bergbreiter, R. Hughes, J. Besinaiz, C. Li and P. L. Osburn, J. Am. Chem. Soc., 2003, 125, 8244–8249 CrossRef CAS.
- D. E. Bergbreiter, J. G. Franchina and B. L. Case, Org. Lett., 2000, 2, 393–395 CrossRef CAS; D. E. Bergbreiter, N. Koshti, J. G. Franchina and J. D. Frels, Angew. Chem., Int. Ed., 2000, 39, 1039–1042 CrossRef.
- A. Datta and H. Plenio, Chem. Commun., 2003, 1504–1505 RSC.
- D. J. Gravert, A. Datta, P. Wentworth, Jr. and K. D. Janda, J. Am. Chem. Soc., 1998, 120, 9481–9495 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn, T. Smith, C. Li and J. D. Frels, J. Am. Chem. Soc., 2003, 125, 6254–6260 CrossRef CAS.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS.
- D. E. Bergbreiter, Macromol. Symp., 1996, 105, 9–16 CAS.
- D. E. Bergbreiter, P. L. Osburn and Y.-S. Liu, J. Am. Chem. Soc., 1999, 121, 9531–9538 CrossRef CAS; A. S. Gruber, D. Zim, G. Ebeling, A. L. Monteiro and J. Dupont, Org. Lett., 2000, 2, 1287–1290 CrossRef CAS; J. Dupont, A. S. Gruber, G. S. Fonseca, A. L. Monteiro, G. Ebeling and R. A. Burrow, Organometallics, 2001, 20, 171–176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details for the synthesis and use of the PIB oligomers and catalysts. See http://www.rsc.org/suppdata/cc/b3/b312368e/ |
| This journal is © The Royal Society of Chemistry 2004 |