The initial stages of aminosilanol polymerisation
Samuel A. French*a, Alexey A. Sokola, C. Richard A. Catlowa, Andreas Kornherrbc and Gerhard Ziffererb
aDFRL, The Royal Institution of Great Britain, 21 Albemarle Street, London, UK W1S 4BS. E-mail: sam@ri.ac.uk; Fax: +44(0)20 7629 3569; Tel: +44(0)20 7409 2992
bInstitute of Physical Chemistry, University of Vienna, Währinger Str. 42, 1090 Wien, Austria
cCenter of Competence in Applied Electrochemistry GmbH, Wiener Neustadt, Austria
First published on 1st December 2003
Abstract
For polysiloxanes to be used as a protective coating it is important that proton transfer, a trigger to polymerisation, is a facile process. Here we investigate the initial stages of polycondensation and compare different silanol tail groups and the effect of solvent (isopropanol). In the case of (3-aminopropyl)trihydroxysilane we see the potential for self catalysis as the tail group is a proton acceptor, while thiolpropyltrihydroxysilane and isopropanol do not promote proton transfer.
The mechanism of the formation of siloxane bonds by polycondensation of silane monomers has been studied extensively1 due to the wide range of technical applications for which polysiloxanes are used, e.g. formation of dense polymer layers on metal substrates, which protects them from corrosion.2 The synthesis of such polysiloxanes normally follows a sol–gel route consisting of various steps (for the sake of simplicity we assume a total hydrolysis of the alkoxy group thus leading to a trihydroxysilane) being catalyzed by some acid or base in an alcoholic solution:
| R′–Si(OR)3 + 3 H2O → R′–Si(OH)3 +3ROH. |
Further polycondensation of the silanol monomers leads to the formation of Si–O–Si bonds and, simultaneously, to the release of water:
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si(OH)
+
(HO)Si
→
Si–O–Si
+ H2O. Si(OH)
+
(HO)Si
→
Si–O–Si
+ H2O. |
As acids and bases act as catalysts, the pH of the solution (along with the monomer and water concentration) plays a crucial role in the formation of polysiloxanes.3 Since the kinetic properties of the system are very complex, theoretical models are an important tool to understand the various intermediates leading to the formation of polysiloxanes. For example, a recent theoretical study4 using ab initio calculations simulated hydrolysis of the siloxane bond under neutral and acidic conditions. So far, no evidence has been provided that the condensation of silanols may proceed in the complete absence of a catalyst.1
However, quantum mechanical (DFT) calculations performed in this study show the possibility that a certain group of organosilanols, i.e. aminosilanols, exhibit self catalysis supporting the condensation process. According to the experimental work of Rousseau et al.5 investigating the polymerisation of (3-aminopropyl)trimethoxysilane (AMO) and ((trimethoxysilane)propyl)diethylenetriamine in methanol solution, there is a weakening of the SiO–H bond by the amino group indicated by a chemical shift in the 29Si NMR spectra.
In this work we investigated this proposal using computational chemistry techniques. To avoid any influence of water we decided to set up a model system consisting of pure hydrolyzed AMO, i.e. (3-aminopropyl)trihydroxysilane (AHO).
No experimental data are available for the structure of an amorphous phase consisting of highly reactive intermediates, like trihydroxysilanes. Therefore, preliminary molecular dynamics (MD) studies were performed to obtain the initial model for the DFT calculations reported below. The starting structure for the MD simulations was built by a modified Markov process (with bond conformational probabilities chosen to account for intramolecular and intermolecular interactions between the molecules) using the Amorphous Cell tool in Materials Studio 2.2.6 We were then able to obtain the density of AHO from NPT MD runs of 200 ps using a large cell of 100 molecules in a cubic box. Periodic boundary conditions were applied with the Anderson barostat under ambient conditions as implemented in Discover, employing the COMPASS forcefield.7 The average value of the density, ρ, over the last 100 ps was calculated and served as the input for the setup of the smaller system of only 10 AHO molecules with a box size of 12.35 Å (this value corresponds to ρ = 1.2104 g cm−3). A subsequent equilibration of this system by a further MD run (NVT, Anderson thermostat) of 100 ps was performed. The final structural configuration from the MD simulation was considered as a well-equilibrated representative of the material and was used as the starting structure for the quantum mechanical DFT calculations.
We employed the same protocol for thiolpropyltrihydroxysilane–THO (10 molecules in a cell with a = 12.73 Å, ρ = 1.2405 g cm−3) and an AHO molecule solvated in isopropanol (1 molecule per 10 solvent molecules, a = 11.58 Å, ρ = 0.7888 g cm−3) used in this work for comparison.
All structures have been modeled at a gradient corrected Density Functional level of theory using the linear combination of atomic orbitals approximation and periodic boundary conditions (employing the PBE exchange-correlation functional and the double numerical with polarisation basis set) as implemented in the DMol3 code.8
We have compared the structural properties of AHO in the gas phase, in solvent and in the bulk. By choosing AHO we are able to introduce a proton acceptor group and compare this with the benign case of THO. The spread of structural motifs across the phases means that we can simultaneously study all the necessary functional groups involved in the different mechanisms of polymerisation that will result from self catalysis and the use of basic promoters.
The starting configuration used in the simulation will bias the structural properties that we observe in the course of our calculations. However, we have the advantage that in the bulk calculations there are ten molecules in different orientations. Therefore, we observe a variety of interactions between silanols in the same simulation cell: head-to-head, head-to-tail and also examples where more than two molecules are in close proximity, forming hydroxyl nests.
Of importance is the nature of the tail group and the solvent as, if they can accept a proton from the silanol head, they can promote self catalysis and provide examples of the mechanisms by which polymerisation occurs. Experimentally, acids or bases catalyse the reaction, and it is therefore impossible to prevent polymerisation once the protector R-groups are removed by hydrolysis from the silanol end of the molecule. In this study, we can effectively freeze the structure in the initial stages of polymerisation during our simulation and compare the effect of different tail groups. We have chosen AHO to illustrate the case of an acceptor tail group with the formation of hydroxyl nests involving, for example three molecules. In contrast, the thiol tail group of THO does not accept a proton. The solvent, isopropanol that we have used in this preliminary study is also benign.
Gas phase AHO—The energy minimised structure of AHO shows that the stability of the molecule is enhanced by the formation of a hydrogen bond, which drives the end groups together leading to a folding of the molecule. The N–C bond length is 1.482 Å, while for the case of the cation there is elongation of the N–C bond to ca. 1.51 Å. For the gas phase anion, the N–C bond length is slightly shorter (1.465 Å) than for the neutral molecule.
Solvated AHO—When surrounded by 10 isopropanol solvent molecules AHO retains the linear structure obtained from the MD simulations (which has also been observed for metastable species in the gas phase). The large steric requirements of moving the solvent before being able to change geometry are the reason that we do not see folding. The lone pair and steric effects associated with the tail group result in a pocket being formed with neither hydrogen attached to nitrogen forming a hydrogen bond and no other hydroxyl coming within 2.8 Å of nitrogen. In contrast, the silanol head forms five hydrogen bonds leading to the elongation of the SiO–H bonds. All solvent molecules form at least one hydrogen bond. The lack of hydrogen bonding to the tail group means the N–C bond length is very similar (1.473 Å) to that of the linear metastable gas phase molecule (1.465 Å).
Bulk AHO—Experimental data from 29Si NMR has suggested that there is interaction between the silanol group and the amine due to the acidic character of AHO. The importance of this interaction is that it will lead to an increase in the SiO–H bond length, which will cause the observed shift to higher field. We would therefore expect this to result in a weakening of the SiO–H bond, which will then facilitate polymerisation and the possibility of self catalysis with the amine group acting as a hydrogen acceptor leading to charge segregation.
In the simulation box, a large number of hydrogen bonds are formed; indeed the final structure is driven by the ability to form these bonds and it is this extensive hydrogen bonding network that allows the amino groups to accept and oxygen to donate protons. We find that there are two anionic and two cationic species in the final structure, while there is one case of a zwitterionic moiety (burgundy – in Fig. 1). We can trace the course of the protons through the simulation, and it is of interest to note that there are only two molecules not involved in proton transfer (magenta and light green). There are three molecules (peach, orange and burgundy), which have two protons that have hopped, as shown in Fig. 1. It must be stressed that in the case where the molecule is in a charged state there is always a very short intermolecular hydrogen bond of the order of 1.7 Å, and all the charged species are in a chain. It is the close intermolecular interactions between molecules that stabilise the proton migration through the bulk.
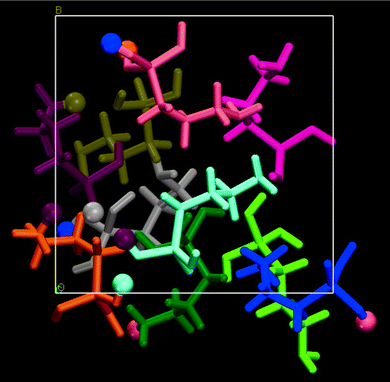 | ||
| Fig. 1 The bulk AHO structure is shown with each molecule being coloured at the beginning of the simulation. Hydrogens that have transferred are shown as balls. | ||
One AHO (grey) can be seen to be curling like a scorpion's tail, with the amino group hydrogen bonding to the silanol head with a N⋯HO distance of 1.622 Å. There are other hydrogen bonds to the head group, which will have aided the folding of the molecule.
The N–C bond length, which varies from 1.477 to 1.499Å, is sensitive to the number of hydrogens coordinated to nitrogen. The longest bond lengths (1.491, 1.495 and 1.499 Å) are all associated with protonated nitrogen. However, it can be seen that the charge is stabilised by the hydrogen bond network as even when NH3+ is present the N–C bond is shorter than that calculated for the gas phase cation. The longest N–C and Si–C (at 1.904 Å) are both found in the zwitterionic molecule.
Bulk thiol—When the tail group is benign to hydrogen donation we observe a network of hydrogen bonds but no proton transfer or hydroxyl nest formation. As for AHO the minimum energy structure shows more hydrogen bonds than the starting structure, as their formation will drive the system to a lower energy confirmation. On the whole the molecules retain the linear structure associated with the gas phase molecule with S–C distances ranging from 1.838 to 1.865 Å, but the average is 1.848 Å, which compares to 1.855 Å for the gas phase molecule.
The calculations performed show that the functional group of the silanol can promote polycondensation. The impact of the functional group is pronounced with protons passing freely between silanol molecules when an acceptor group is present.
We thank the Royal Society for the financial support, EPSRC for the computational resources and Accelrys for the software. Furthermore, AK is also grateful to TIG and the government of Lower Austria for financial support within the Kplus program.
Notes and references
- (a) W. Noll, The Chemistry and Technology of Silicons, Academic Press, New York, 1968 Search PubMed; (b) J. Sjöblom, G. Stakkestad and H. Ebeltoft, Langmuir, 1995, 11, 2652 CrossRef; (c) F. D. Osterholz and E. R. Pohl, J. Adhesion Science and Technology, 1992, 6, 127 Search PubMed.
- A. M. Beccaria and L. Chiaruttini, Corrosion Science, 1999, 41, 885 Search PubMed.
- L. Wilczek and J. Chojnowski, Makromol. Chem., 1983, 184, 77 CrossRef CAS.
- M. Cypryk and Y. Apeloig, Organometallics, 2002, 21, 2165 CrossRef CAS.
- F. Rousseau, C. Poinsignon, J. Garcia and M. Popall, Chem. Mater., 1995, 7, 828 CrossRef CAS.
- Materials Studio 2.2. Accelrys Inc. 2002.
- (a) H. Sun, Macromolecules, 1995, 28, 701 CrossRef CAS; (b) H. Sun, J. Phys. Chem. B, 1998, 102, 7338 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 3865 CrossRef CAS; (b) B. Delley, J. Chem. Phys., 1990, 92, 508 CrossRef CAS; (c) B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |