An endothelial and astrocyte co-culture model of the blood–brain barrier utilizing an ultra-thin, nanofabricated silicon nitride membrane
Sarina Harris Maa, Lori A. Lepakb, Rifat J. Hussainc, William Shainc and Michael L. Shuler*a
aCornell University Dept. of Chemical and Biomolecular Engineering, 270 Olin Hall, Ithaca, NY, USA 14853. E-mail: mls@cheme.cornell.edu; Fax: (607) 255-9166; Tel: (607) 255-7577
bCornell University, Dept. of Chemistry and Chemical Biology, Ithaca, NY, USA
cLaboratory of Nervous System Disorders, Wadsworth Center, Albany, NY, USA
First published on 14th October 2004
Abstract
The endothelial cells comprising brain capillaries have extremely tight intercellular junctions which form an essentially impermeable barrier to passive transport of water soluble molecules between the blood and brain. Several in vitro models of the blood–brain barrier (BBB) have been studied, most utilizing commercially available polymer membranes affixed to plastic inserts. There is mounting evidence that direct contact between endothelial cells and astrocytes, another cell type found to have intimate interaction with the brain side of BBB capillaries, is at least partially responsible for the development of the tight intercellular junctions between BBB endothelial cells. However, the membranes commonly used for BBB in vitro models are lacking certain attributes that would permit a high degree of direct contact between astrocytes and endothelial cells cultured on opposing sides. This work is based on the hypothesis that co-culturing endothelial and astrocyte cells on opposite sides of an ultra-thin, highly porous membrane will allow for increased direct interaction between the two cell types and therefore result in a better model of the BBB. We used standard nanofabrication techniques to make membranes from low-stress silicon nitride that are at least an order of magnitude thinner and at least two times more porous than commercial membrane inserts. An experimental survey of pore sizes for the silicon nitride membranes suggested pores ∼400 nm in diameter are adequate for restricting astrocyte cell bodies to the seeded side while allowing astrocyte processes to pass through the pores and interact with endothelial cells on the opposite side. The inclusion of a spun-on, cross-linked collagen membrane allowed for astrocyte attachment and culture on the membranes for over two weeks. Astrocytes and endothelial cells displayed markers specific to their cell types when grown on the silicon nitride membranes. The transendothelial electrical resistances, a measure of barrier tightness, of endothelial and astrocyte co-cultures on the silicon nitride membranes were comparable to the commercial membranes, but neither system showed synergy between the two cell types in forming a tighter barrier. This lack of synergy may have been due to the loss of ability of commercially available primary bovine brain microvascular endothelial cells to respond to astrocyte differentiating signals.
Introduction
The endothelial cells lining brain capillaries form an almost impermeable barrier to water soluble molecules lacking a specific transporter between the blood and the brain. This blood–brain barrier (BBB) is the result of complexes of proteins located at cell–cell contacts called tight junctions which work to seal adjacent membranes together.1 The BBB protects the brain and provides a constant environment for neural processes. Although it is still not clear exactly which chemical signals (and their timing and source) are involved in the specialization of the BBB endothelial cells, astrocytes (a type of microglial cell) appear to play a role in this differentiation.2–4Several in vitro models of the BBB have been developed with the aim to better understand the cell biology of the BBB and/or to predict potential therapeutic permeability into the brain (see for reviews refs. 5–9). Most of these models consist of a monolayer of endothelial cells, often isolated from brain capillaries of animals, grown on the top side of a polymeric membrane affixed to a cylindrical plastic insert. This culture insert is placed into a well of a standard culture plate, dividing the well into luminal (blood) and abluminal (brain) chambers. The barrier properties of the model are usually assessed by one or more of the following: transendothelial electrical resistance (TER) measurements, direct determination of molecular permeability coefficients, immunofluorescent labeling of proteins (such as occludin and ZO-1) specific to tight junctions, and other techniques to detect proteins found to be highly expressed at the BBB (such as P-glycoprotein and γ-glutamyl transpeptidase).
Unfortunately, once endothelial cells are isolated from brain capillaries, they begin to lose their BBB characteristics quickly when cultured alone. Several groups have attempted to better recreate the brain environment by incorporating astrocyte or astroglial co-cultures into their in vitro BBB models (see for reviews refs. 5–9). The co-cultures are achieved either by using an astrocyte conditioned medium, by growing the astrocytes on the bottom of the well in which the cell culture insert is placed, or by seeding astrocytes directly on the bottom of the insert membrane. These co-cultures have been shown to increase the barrier properties of the model. However, there is still debate as to whether direct contact between the endothelial and astrocyte cells is required for more complete endothelial cell differentiation or if some factor(s) secreted by astrocytes are sufficient.2,3 The models with the two cell types cultured on opposite sides of the membrane best recreate the anatomy of the BBB in which astrocyte foot processes show a high degree of direct interaction with brain capillaries.3
Results from several in vitro BBB models support the hypothesis that direct contact is necessary for full BBB endothelial cell differentiation.10–12 However, the membranes in the commercially available culture inserts do not have physical dimensions which promote a high degree of interaction between two cell types grown on opposing sides. These membranes are 10 µm thick, come in limited pore sizes (0.4 µm, 1.0 µm, and 3.0 µm), have low to medium porosities (maximum 15%), and are not transparent for easy microscopic observation of cultured cells. The track-etching process used to produce most of these membranes also results in a fairly wide pore-size distribution and a random arrangement of pores.
The work presented here is based on the hypothesis that increasing the amount of direct contact between endothelial cells and astrocytes while maintaining two distinct cell layers will lead to a more differentiated and appropriate BBB model. We tested this hypothesis by utilizing membranes nanofabricated from low-stress silicon nitride (SiN). These membranes are at least an order of magnitude thinner than the commercially available membrane culture inserts, have a very narrow pore size distribution and well controlled pore arrangement, and can have porosities approaching 50%. These attributes should allow for greater interaction between endothelial and astrocyte cells grown on opposing sides of the fabricated SiN membranes in comparison to the commercial membranes. We have previously reported our initial efforts to develop these membranes for endothelial cell culture.13 Here we describe the development of these fabricated membranes for endothelial and astrocyte co-cultures.
Materials and methods
Materials
The source of some materials has been reported previously.13 These additional materials were purchased from the indicated vendors: 897-12i photoresist from OCG Microelectronics; ZEP-520 e-beam resist from Zeon Corporation (Tokyo, Japan); Falcon polyethylene terephthalate (PET) Transwell membrane inserts for 24-well plates and Falcon 24-well plates for Transwell membrane inserts from Fisher Scientific (Pittsburgh, PA); molecular biology grade bovine skin collagen type I from US Biological (Swampscott, MA); 1 mm thick cell culture grade silicone from Grace BioLabs (Portland, OR); glutaraldehyde solution, paraformaldehyde, sodium cacodylate buffer, osmium tetraoxide, and uranyl acetate from Electron Microscopy Sciences (Ft. Washington, PA); rat tail collagen type I and monoclonal anti-E-cadherin mouse IgG2a antibody from BD Biosciences (Bedford, MA); monoclonal anti-collagen type I mouse IgG1 antibody, FITC conjugated anti-rabbit goat IgG, 100x antibiotic–antimycotic solution, and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) from Sigma (St. Louis, MO); monoclonal anti-GFAP mouse IgG1 antibody from Chemicon Intenational (Temecula, CA); polyclonal anti-occludin rabbit antiserum from Zymed (S.San Francisco, CA); Rhodamine Red™-X- conjugated AffiniPure goat anti-mouse IgG from Jackson ImmunoResearch (West Grove, PA); acetylated low density lipoprotein labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-carbocyanine percholorate (DiI-Ac-LDL) from Biomedical Technologies (Stoughton, MA); Vectashield mounting medium from Vector Laboratories (Burlingame, CA).Fabrication of silicon nitride membranes
SiN membranes were fabricated by a procedure similar to that outlined in Kuiper et al.,14 with the difference that the through wafer KOH etch to create unpatterned silicon nitride windows and the lithography to create the pattern of pores in the silicon nitride window were done in separate sequential steps. Also, we used either a lithographic stepper or an electron beam lithography system to pattern the pores. The patterning of the silicon nitride windows is discussed in detail elsewhere.13 Briefly, the backs of standard 100 mm , <100>, single-side polished silicon wafers with 1 µm thick low pressure chemical vapor deposition low-stress silicon nitride films on each side were patterned with the Hybrid Technology Group's 3HR contact/proximity mask aligner. The mask was designed to divide the wafer into 24 chips, each 15 mm × 15 mm and centered on a single 1500 µm × 1500 µm square. The patterned backside SiN layer was plasma etched to the silicon underneath and SiN windows created by a through wafer etch in 15% (w/v) KOH in water at 85 °C. As the side-walls of a KOH etch form a 54.5° angle with the wafer surface, the final dimensions of the SiN windows on the top side were approximately 800 µm × 800 µm × 1 µm thick as shown in Fig. 1. There was some variation in the window size between wafers due to slight misalignments with the wafer crystal planes. We will refer to the volume of silicon etched below the membrane as the membrane well.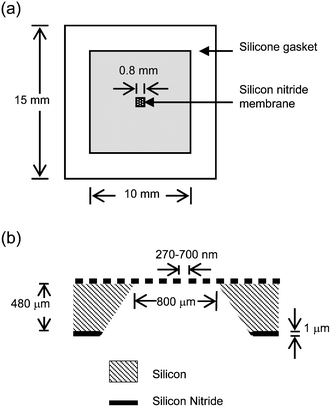 | ||
| Fig. 1 Schematics of chip and membrane layout. (a) Top view of chip and gasket assembly. (b) Cross section of the silicon nitride membrane after fabrication (not to scale). | ||
The procedure for patterning the array of 700 nm pores is nearly identical to the procedure for patterning larger pores as described elsewhere.13 Briefly, the wafer with silicon nitride windows was primed with P20 and then spun with 1.3 µm of OCG 897-12i photoresist which was pre- and post-exposure baked for 1 min on a 90 °C hotplate. A mask was designed to expose a regular array of 600 nm diameter pores with a 1.2 µm period and rectangular packing over the entire area of the silicon nitride window using a GCA 10x i-line stepper. The pattern was developed in MIF300 for 1 min. The photoresist was then used as an etch mask to transfer the pore pattern through the nitride window using a PlasmaTherm72 etcher with a recipe of 40 mTorr pressure, 150 W power, and 29.4 sccm CF4.
Pores 300–500 nm were directly written onto the nitride windows using a Leica/Cambridge EBMF 10.5. The top sides of wafers patterned with SiN windows were cleaned with acetone and then isopropyl alcohol on a wafer spinner with a vacuum chuck. ZEP 520 resist was spun to 500 nm thick on the top and then baked for 2 min at 170 °C. The EBMF was programmed to expose regular hexagonal arrays of various sized pores, one pore size per SiN window. The ZEP 520 was then used as an etch mask to transfer the pore pattern through the nitride window using an Oxford PlasmaLab 80+ RIE system with a recipe of 55 mTorr pressure, 150 W power, 50 sccm CHF3, and 5 sccm O2. This recipe gave better resist to nitride selectivity compared to the CF4 etch recipe. However, the high aspect ratio for etching 300–500 nm pores into 1 µm of low-stress silicon nitride slowed the SiN etch rate in the pores, and the resist did not protect the unpatterned portions of the nitride for the entire etch. The typical resulting membranes were 500 nm thick with 300–500 nm pores and porosities of 20–50%. The through SiN pore size was confirmed by a Zeiss 982 scanning electron microscopy (SEM) of the back side of the patterned silicon nitride windows.
Chip cleaning and preparation
Wafers with patterned silicon nitride windows were broken along dice lines into 1.5 cm × 1.5 cm chips with a single membrane in the center. Chips were cleaned first with 4% ammonium hydroxide with 4.3% hydrogen peroxide at 80 °C for 5 min each side. After two washes in distilled water, chips were placed in a 12.5% sulfuric acid and 4.3% hydrogen peroxide solution at 80 °C for 5 min each side. One mm thick cell culture grade silicone was cut into 1.5 cm × 1.5 cm squares with a 1 cm × 1 cm square removed from the center to form a gasket. The silicone gasket was treated in the same sulfuric acid/hydrogen peroxide solution as the chips for 5 min on each side. The chips and silicone gaskets were put through a series of six rinses in distilled, deionized (DI) water. The wet gaskets were then centered on the top side of the wet chips, excess water was removed from the square inside the gasket, and the two were allowed to dry together either overnight at room temperature or for 30 min to 1 h in a 60 °C oven. We will refer to the volume inside the silicone gasket as the chip well. A second wet gasket was then placed on the bottom side of the chip by first wetting the chip with DI water, centering the gasket, removing excess water, and drying as for the top side gasket. If the chip was spun with collagen, then the gasket was placed on the bottom side after the collagen spinning.Collagen membrane application
The backsides of the chips were first primed with glutaraldehyde vapor at 4 °C for 1 h. The chip was placed bottom side up into a vacuumless wafer spinner chuck made in-house with a recess cut in the center the length and depth of the square chip. A few drops of 0.3% bovine skin collagen type I solution were spun onto the wafer for 1 min at 2500 rpm with a 10 s ramp up and down time. This was repeated a second time. If the chips were to be used with bovine brain microvascular endothelial cells (bMVEC-B), then two layers of collagen were also spun as described on the top side of the chip. The chip was rinsed ∼1 s in running DI water and placed into a 0.02% solution of glutaraldehyde for 1 h at 4 °C. The chips were then rinsed in DI water and allowed to dry at room temperature. The bottom side silicone gasket was attached as described in the previous section.Astrocyte isolation and cell culture maintenance
Astrocytes were isolated from 3 day old Wistar rat pups following established techniques.15 Cells were maintained in Dulbecco's modified Eagle medium (DMEM) from Invitrogen (Carlsbad, CA) supplemented with 5% fetal bovine serum and antibiotic–antimycotic solution at the supplier's recommended concentration. The cultures were fed every 3–4 days by completely replacing the medium. Seventy percent confluent cultures were gradually frozen to −80 °C in cell culture medium with 10% dimethylsulfoxide and then transferred to liquid nitrogen for storage. Vials of frozen astrocytes were quickly thawed in a 37 °C waterbath, the contents added to 5 ml culture medium, and the cells spun from solution at 70 g for 5 min. The pelleted cells were suspended in 5 ml fresh medium and transferred to a 25 cm2 tissue culture flask. Astrocytes were typically used for experiments 14–21 days post thawing. Occasionally first passage astrocytes were used for experiments if they tested positive for GFAP, a common astrocyte marker.The immortalized brain capillary endothelial cell line SV-HCEC was kindly provided by A. Muruganandam and D. Stanimirovic of the National Research Council of Canada.16 Although this cell line is reported to be of human origin, we were alerted (personal communication, Françoise Roux, Unité de Neuro-Pharmaco-Nutrition Hôpital Fernand Widalk, France) to probable contamination of the cell line with cells of rat origin. Karyotype analysis and negative labeling with an anti-human nuclei antibody (Chemicon mAb 1281) confirmed the rat origin of this cell line. We learned of this discrepancy after the experiments shown in Fig. 3 (see later) were completed. However, the cell line does have distinct endothelial morphology, which should be adequate for interpretation of the results shown in Fig. 3. The SV-HCEC line was cultured in M199 medium from Invitrogen with 10% FBS, 1× insulin–transferrin–selenium, and heparin at 600 USP units L−1. Every 3 to 4 days cells were either fed with fresh medium or split 1∶20 if nearly confluent.
Cryopreserved, single donor, human umbilical vein endothelial cells (HUVEC), M200, trypsin, and trypsin neutralizing solution were purchased from Cascade Biologics (Portland, OR). HUVEC were thawed and maintained in M200 medium according to the supplier's recommendations. Cryopreserved, single donor, bovine aortic endothelial cells (BAEC), cryopreserved pooled bovine brain microvascular endothelial cells (bMVEC-B), EGM-MV Bulletkit, EMVB Bulletkit, and ReagentPack subculture reagents were purchased from Cambrex Bio Science (Walkersville, MD). Again, the BAEC and bMVEC-B were thawed and maintained according to the supplier's recommendations. All cultures were maintained in a 37 °C humidified cell culture incubator with 5% CO2. Cells were observed with an Olympus inverted phase contrast microscope and digitally captured with a Sony 3CCD Ex wave HAD color video camera.
Cell seeding of fabricated membranes
All solutions and plastic-ware used for tissue culture were sterile unless indicated otherwise, and cell seeding was done in a biosafety tissue culture hood using aseptic technique. If the chip did not have a spun-on collagen membrane, it was autoclaved and then placed top side up onto a 100 µl drop of either 60 µg ml−1 human plasma fibronectin in phosphate buffered saline (PBS), 0.1% gelatin in water, 100 µg ml−1 collagen in 0.02 N acetic acid, or 100 µg ml−1 polylysine in water in a Petri dish. Chips were visually inspected to ensure that no air bubbles were trapped in the bottom chip well. One hundred microliters of 60 µg ml−1 human plasma fibronectin in PBS were then pipetted into the top chip well. The chips were placed into a 37 °C humidified cell culture incubator for 1 h. The protein/amino acid solutions were aspirated from the chips, and the chips were rinsed once in culture medium and placed into 1 ml cell culture medium in a well of a 6-well culture plate. The chip side which was facing up depended on whether the astrocyte or endothelial cells were to be seeded first. We tried both seeding orders and obtained the best results if the endothelial cells were seeded first and allowed to establish a confluent monolayer before the astrocytes were seeded on the chip bottom.If a collagen membrane was spun onto the chip, the chip was not autoclaved. To sterilize the chip and to wet the membrane well, the chips were placed bottom up into 70% ethanol in a Petri dish for 10 min, then transferred to a second Petri dish with cell culture medium containing antibiotic–antimycotic at three times the supplier's recommended concentration for 10 min, rinsed in a third dish containing standard cell culture medium for 10 min, and finally moved into 1 ml of cell culture medium in a 6-well plate with the top side up. Into the chip well, 60 µg ml−1 of fibronectin solution was pipetted and allowed to absorb at 37 °C for 1 h. The protein solution was aspirated, and the chip top side was rinsed once with cell culture medium.
The protocol for cell seeding onto the SiN membranes was the same for all protein preparations. Endothelial cells were trypsinized from the culture flask using the cell suppliers' recommended trypsin solution, suspended in trypsin neutralizing solution, pelleted from solution by a 5 min spin at 70 g, and resuspended in cell culture medium. The density of cells in suspension was determined with a hemacytometer using 50% trypan blue, and the endothelial cell suspension was adjusted to 60 × 104 cells ml−1. A 100 µl volume of the cell suspension was placed in the top chip well for a seeding density of 6 × 104 cells cm−2. The endothelial cells were allowed to attach for either 8 h or overnight except for bMVEC-B cells which were allowed to attach for 24–36 h. The medium in the top chip well was then aspirated to remove any non-attached cells, and 1 ml of medium was added to the well if a second cell type was not to be seeded immediately. The astrocyte seeding procedure was similar, except that the trypsin came from Life Technologies (Rockville, MD), cell culture medium was used to neutralize the trypsin, and the cells were seeded at a density of 8 × 103cells ml−1. The astrocytes were plated in the appropriate cell culture medium for the endothelial cells used in the experiment. Each endothelial medium was pre-tested for its ability to support astrocyte attachment and proliferation. Every other day, 1 ml of medium was removed from each well and replaced with 1 ml of fresh culture medium. The chips were also flipped each day so the cells spent equal time facing either up or down.
Seeding of commercial membranes
For all experiments with commercial membranes we used Falcon PET membranes for 24-well plates. Due to the adhesion site of the membrane to the plastic support, these membranes have a topside surface area of 0.3 cm2 and an approximate backside surface area of 0.6 cm2. The top sides of Falcon PET membranes for 24-well plates were first treated with 50 µl of 100 µg ml−1 collagen in 0.02 N acetic acid at 37 °C for 1 h only if they were to be seeded with bMVEC-B. The collagen solution was aspirated from the membranes and the membranes rinsed once with 100 µl PBS. Regardless of which endothelial cells were to be seeded, the membrane top side was treated with 100 µl of 20 µg ml−1 human plasma fibronectin in PBS for 1 h at 37 °C. The membranes were rinsed once with 100 µl PBS and flipped upside down in a tall Petri dish. A 45 µl volume of 16 × 104 cells ml−1 astrocyte cell suspension, prepared as for the SiN membrane seeding, was pipetted onto the membrane backside. The membranes were placed in the cell culture incubator for ∼25 min to allow for cell attachment. The membranes were flipped right side up into 700 µl of cell culture medium. One-hundred microliters of medium was added to membranes to be seeded with endothelial cells later and 200 µl to membranes not being seeded with endothelial cells. The astrocytes were allowed to attach for either 8 h or overnight before endothelial cell seeding. A 100 µl volume of 20 × 104 cells ml−1 endothelial cell suspension was then added to the topside. The commercial membranes were fed every other day by complete removal and replacement of the cell culture medium on both sides of the membrane.TER measurement
TER measurements of the cultures were made every 3 days after both endothelial cells and astrocytes had been seeded. TER measurements of the cells on the Falcon PET membranes were made using the Millicell ERS from Millipore (Billerica, MA) with the Endohm-6 chamber from World Precision Instruments (Sarasota, FL). The chamber was sterilized prior to use with 70% ethanol for 15 min. The cup was rinsed once with 700 µl of measurement medium. The measurement medium for TER always consisted of the cell culture medium without serum, as serum proteins tended to deposit on the electrodes. The Endohm chamber was equilibrated with 700 µl of measurement medium for ∼1 h at room temperature. Before taking measurements, the Millicell ERS voltage was adjusted to a zero reading and the equilibrating medium was replaced with 700 µl fresh measurement medium. The medium in the tissue culture wells was allowed to come to room temperature by removing the plates from the incubator 5 min prior to measurement. Individual membranes were removed from the well plate and placed into the Endohm-6 cup for TER measurement. Two measurements at different membrane insert positions were taken per sample.A chamber for measuring the TER of the fabricated SiN membranes was made in-house as shown in Fig. 2. The device consisted of 2 pieces of 1/4″ thick Lexan. Into each piece of Lexan a first recess (27.5 mm × 15.5 mm) was cut, 0.6 mm and 1.2 mm deep in the top and bottom faces respectively. A second recess was cut in the center of the first recess with dimensions of 25 mm × 10 mm. The second recess was 3 mm deep measured from the original Lexan face. The first, shallower recess served as a ledge for the silicone gaskets, and the second, deeper recess formed a media reservoir between the sides of the chips once the two faces were screwed together. Holes for 2 mm diameter × 4 mm length silver/silver chloride electrodes (World Precision Instruments) were drilled in the deeper recess. The electrodes were sealed into the chamber with clear epoxy placed on the outside of the chamber only. A set of four holes were also drilled at the corners of the device for stainless steel feet (bottom face)/guide holes (top face) and another set of four countersunk holes closer to the center of the Lexan sheets for 6–32 flathead screws. Two small copper plates were fixed with two 2–56 screws at one side on each piece of Lexan. The wire of the electrode was screwed into one end of the copper plate and the corresponding lead to the Millicell ERS into the other end.
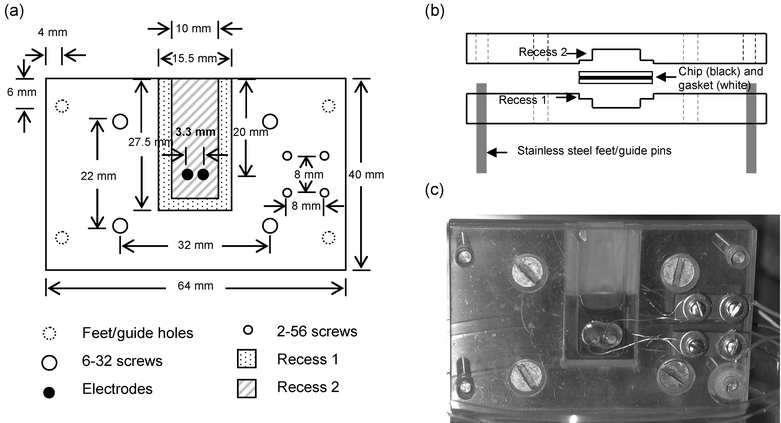 | ||
| Fig. 2 Schematics and pictures of the TER measurement device for fabricated membranes. (a) Top view schematic of the chamber with dimensions and a legend. (b) Side view of the chamber assembly including the silicon nitride chip with gaskets and the chamber profile when viewed from the top edge of Fig. 2(a). (c) Photograph of the device from the top view shown in (a) (sitting in a plastic Petri dish) in use. | ||
Before taking measurements, the opened chamber was sterilized by placing the recessed sides face down into 70% ethanol for 15 min along with submerging the screws and a dummy chip with silicone gaskets. The non-recessed faces of both sides of the chamber were not immersed in the ethanol. The chamber was air dried in the culture hood before use. To measure the TER of the fabricated membrane, the chip was removed from the 6-well plate and placed backside up into the bottom recess. The guide pins were used to position the top piece of the resistance chamber into place. If chip alignment was satisfactory, the four screws were alternately tightened around the chip, and the silicone gaskets separated the chip sides into two chambers. Measurement medium (275 µl and 250 µl) was added to the chip back side and front side chambers respectively. Before taking sample measurements, the electrodes were equilibrated with measurement medium for 1 h using the dummy chip. The measurement was taken with the Millicell ERS, the medium was removed from each side of the chip using a glass Pasteur pipet, the device was unscrewed, and the chip was placed back into the 6-well plate. Any variations in membrane size resulting from misalignment with the wafer crystal planes for the KOH etch were taken into account by measuring the actual size for each wafer and only using chips from the same wafer for a single experiment.
SEM of cells on membranes
The silicone gaskets were removed from the chips before fixation and all steps were done top side up. Unless otherwise noted, the fixation buffer was 0.1 M sodium cacodylate in water, pH 7.4. Cells on the chips were fixed by first rinsing once with room temperature PBS and then placing in 2.5% glutaraldehyde in fixation buffer for 1 h at room temperature and 30 min on ice. The chips were rinsed three times for 5 min each with fixation buffer on ice and then placed into 2% osmium tetroxide in fixation buffer on ice for 1 h. The chips were rinsed three times with fixation buffer on ice as before. The cells were then dehydrated through a series of 10%, 30%, 50%, 70%, 90% and 100% solutions of ethanol in water for 10 min each except for the 70% solution which also included 2% uranyl acetate for a 20 min incubation. The first three ethanol stages were done on ice and the rest at room temperature. The chips were critically point dried with ethanol and CO2. Chips were mounted onto large SEM stubs with double sticky tape and then sputter coated with ∼30 nm Au/Pd. The fixed cells were viewed using a Hitachi S4500 SEM at 5 kV accelerating voltage.Immunofluorescent labeling
The cells were rinsed once with PBS. If the cells were to be labeled for occludin, they were pre-permeabilized with 0.2% Triton x-100 in PBS for 2 min on ice and then rinsed once with PBS. All cells were fixed in 3.7% paraformaldehyde in PBS for 15 min at room temperature and permeabilized with 0.05% Triton x-100 in PBS for 5 min at room temperature. The cells were then rinsed once in PBS at room temperature and then rinsed twice with 10 min between rinses with PBS containing 2% goat serum to block non-specific binding. When labeling just the collagen membranes, no fixation or permeabilization was done and the protocol was started at the previous rinse step. The chip was positioned with the cells to be labeled facing up in 1 ml PBS. The following dilutions in PBS were used for the primary antibodies: anti-collagen 1∶10, anti-GFAP 1∶50, and anti-occludin 1∶25. To the appropriate chip well, 100 µl of the diluted primary antibody(s) was added for 1 h in a 37 °C humidified incubator or overnight at 4 °C in a sealed chamber with wetted tissues to provide a humidified environment. The cells were rinsed three times for 10 min each with PBS containing 2% goat serum and then incubated 30 min in a 37 °C incubator with the appropriate fluorescently labeled secondary antibody(s) diluted 1∶50 in PBS. The cells were sometimes rinsed once with PBS with 2.5 µg ml−1 DAPI for 10 min to observe cell nuclei. The labeling was finished by three rinses in PBS. Pictures were either taken immediately, or the specimens were mounted on a glass slide with a coverslip in Vectashield mounting medium for later analysis. The antibody labels were viewed with an Olympus BX51WI upright microscope fit with the appropriate wavelength filter cubes and digital images were captured using a Retiga 1300 CCD camera.Uptake of DiI-Ac-LDL
DiI-Ac-LDL was diluted to 10 µg ml−1 in the growth medium appropriate for the endothelial cells. One milliliter of medium was removed from the culture plate well containing the chip(s) of interest. If necessary, the chip was flipped endothelial side up and any culture medium in the chip well was aspirated. The cells were incubated at 37 °C for 4 h with 100 µl of the diluted DiI-Ac-LDL pipeted into the chip well. The cells were washed three times in PBS and fixed for 15 min in 3.7% paraformaldehyde in PBS at room temperature. The chips were rinsed twice with PBS and either viewed immediately using a rhodamine fluorescence cube or mounted in Vectashield for later analysis.Results and discussion
Selection of membrane pore size
We did an initial experiment to determine whether endothelial and astrocyte cells could be either plated together from a mixed cell suspension and then reorganize into two distinct cell layers or plated one layer after another on the same side of the membrane and then maintain two distinct cell layers. The results of this experiment are shown in Fig. 3. The first two bars show TERs for the astrocyte and endothelial layers alone, respectively. For two cell layers (resistors) in series, we would expect the resistance to be equal to the sum of the two monolayer resistances if there is no synergy between the cells. However, from this graph, we can see if the cells are directly plated on the same membrane side, either in a mixed cell suspension or by allowing establishment of the first layer before plating the second, the resulting resistance of the two cells is similar to either the astrocyte or endothelial cell layer alone. This result indicates that two distinct cell layers did not form. In comparison, if the astrocytes are plated on the backside of the membrane and the endothelial cells plated on the topside of the membrane, the resulting resistance is equal to the sum of the astrocytes and endothelial layer resistance within experimental error. Based on this observation, a membrane is necessary to separate the cell bodies of the two cell types and form a more realistic BBB model.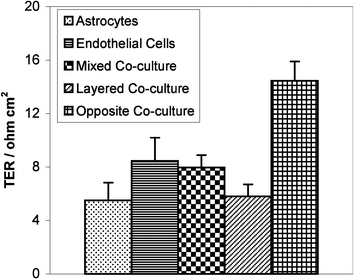 | ||
| Fig. 3 A comparison of different co-culture methods of rat astrocytes and the SV-HCEC line on 1.0 µm pore size commercial membrane inserts. The first two bars are for astrocytes and endothelial cell line layers alone. For the mixed co-culture, the astrocytes and endothelial cells were mixed in suspension and plated together on the membrane topside, whereas for the layered co-culture, the astrocytes were allowed to attach for 6 h before plating the endothelial cells directly on top. Astrocytes were allowed to attach to the bottom of the membrane for 8 h before endothelial cells were plated on the top of the membrane for the opposite co-culture. Measurements are shown 6 days post cell seeding and error bars are +1 standard error. | ||
The results of the initial experiment imply that it would be important to design the fabricated membrane with a pore size that would allow growth through of the astrocyte foot processes while restricting the astrocyte and endothelial cell bodies to the seeded side. SEM pictures of astrocyte cell growth through commercial membranes of 3.0 µm, 1.0 µm, and 0.4 µm show that primary astrocyte cell bodies readily grow through 3.0 µm pores, extend foot processes through 1.0 µm pores, and do not grow through 0.4 µm pores (personal unpublished results and ref. 10,17). We initially designed a mask to fabricate pores of 1.2 µm or 0.6 µm depending if a 5× or 10× stepper was used. After the plasma etch through 1 µm of silicon nitride, we typically had pores with sloped sidewalls measuring 750 nm from the patterned (top side) and 600 nm from the backside if the 10× stepper was used. Unless otherwise indicated, we will indicate pore sizes as the smaller of the two dimensions, as this is the dimension the astrocyte processes must actually traverse. The pore size was very uniform across a single membrane, but there was a small variation in pore size across a wafer (typically ∼5%) with the membranes closer to the wafer edge having slightly larger pores than those at the center. We found this variation not to cause significant differences between TER measurements of blank fabricated membranes. However, variation in pore size between wafers was larger (up to ∼15%) than between chips on a single wafer. To reduce the effects of this between wafer variation, only membranes from the same wafer were used in a single experiment. Early endothelial and astrocyte co-culture results on the fabricated membranes with 600 nm pores showed resistances similar to the astrocytes cultured alone on the fabricated membrane. SEM of the side opposite to the cell seeded side of fabricated membranes seeded only with astrocytes showed substantial growth through the membrane of entire astrocyte cell bodies. It was not surprising that the pore size to restrict astrocyte cell bodies to the seeded side of the membrane would be smaller for the fabricated SiN membranes compared to the thicker, less porous commercial membranes.
In order to restrict the astrocyte cell bodies to the seeded side, we did a study with a series of progressively smaller pores. Because the 600 nm pores were already near the size limits of the available stepper technology, we used electron beam lithography to direct write the pattern of pores. The pore packing was changed to a hexagonal array to improve the membrane strength. For prototype purposes, we used 500 nm of ZEP 520 e-beam resist as the etch mask. However, the etch selectivity of the SiN to the resist was not high enough to withstand etching of 300–500 nm pores through 1 µm of SiN, and the membranes were typically 500 nm thick once the pores were etched through. Again, the pores had sloped side-walls with a difference of ∼75 nm diameter from the top to the bottom. We fabricated two sets of membranes with pore sizes of 290 nm and 420 nm. The topography of the resulting fabricated membranes can be compared in Fig. 4. In all cases, the pores are regularly spaced, with hexagonal and rectangular packing for the two smaller and largest pores respectively. A difference in porosities between the different pore sizes can be observed, which was due to the poor etch resistance of the electron beam resist.
 | ||
| Fig. 4 SEM images of the side opposite to astrocyte seeding on three membrane pore sizes: (a) 290 nm, (b) 420 nm, (c) 600 nm. Cells were able to readily grow through the 600 nm pores, one to two cells per membrane grew through 420 nm pores, and no cells were able to get though the 290 nm pores. This figure also illustrates the different morphologies of the astrocyte extensions that grew through the pores. | ||
Astrocytes were grown on the backside of the different pore sized membranes and SEM was use to probe membranes for cell growth through the membrane. Examples of the qualitative results of this study are illustrated in Fig. 4, which show the membrane side opposite to the side on which astrocytes were seeded. We observed that several cells per membrane were able to easily traverse the 600 nm pores, only one to two cells per membrane crossed through the 420 nm pores, and no cells were able to grow through the 290 nm pores. Quantitative results from this study were complicated by cell division; it was not possible to determine in a group of cells which had grown through the membrane and which were daughters of a cell that had previously grown through. Also illustrated in Fig. 4 are the different morphologies the astrocyte extensions which grew through the membrane displayed. There were round extensions (a), long “stick-like” extensions (b), and flat, almost cell body like, but smaller, extensions (c). Despite complications in quantifying the results of the pore size study, the qualitative results led us to target membranes with a pore size 350–400 nm. Unfortunately, due to the quick radial etch rate of the pores once the photoresist was gone, we were not able to precisely control the pore size using a soft etch mask and typically ended with pores ∼420 nm. The use of a hard mask with high etch resistance to CHF3 and CF4 reactive ion plasmas should improve control over membrane pore size, allow for thicker (∼1 µm) membranes, and possibly reduce the side-wall slope.
Collagen membrane for astrocyte culture
Silicon nitride has been found to be non-toxic to rat brain slices18 and to allow proliferation of and not promote an inflammatory response in a human osteoblast cell line.19 Low-stress silicon nitride, which is silicon enriched compared to a stoichiometric Si3N4 film, supported growth of a fibroblast cell line.20 To our knowledge there are no previously published studies of astrocyte attachment and long-term culture on silicon nitride. When the fibronectin treatment routine was developed for the endothelial cells,13 we assumed that the astrocytes would adhere to and divide on the same treated surface. However, the astrocytes did not stay attached to the SiN membranes treated with fibronectin and would tend to “ball up” after a few days in culture. This detachment was especially prevalent when the cells were temporarily removed from their medium environment for TER measurements. Absorbing polylysine at 10 µg cm−2 to the membrane bottom side in place of the fibronectin also resulted in astrocyte detachment. Originally we thought this detachment might be caused by the sharp angle at the interface between the SiN and silicon at the bottom of the membrane well resulting from the KOH etch. However, when we compared astrocyte growth on non-patterned SiN membrane wells to growth on silicon membrane wells, created by stopping the KOH etch before reaching the SiN, the astrocytes did not detach from the silicon well, but did detach from the silicon nitride well (unpublished results). The astrocytes also tended to stay attached to the smooth silicon <111> sidewalls in the fully etched membrane wells even when they detached from the SiN membrane (unpublished results). These observations indicated that the fibronectin treated SiN material itself was at least partially responsible for the poor astrocyte attachment. The fibronectin results are consistent with a recent study showing that astrocytes do not show increased attachment to glass modified with fibronectin compared to the control.21 It has also been shown that astrocyte adhesion tends to decrease with increasing contact angle (i.e. increasing hydrophobicity),22 and we found that fibronectin adsorption significantly increases the hydrophobicity of the silicon nitride surface.13 However, these results do not address the failure of the poly-lysine coating, which typically promotes cell attachment by increasing electrostatic attraction between the cell and a surface. It was previously observed that juvenile rainbow trout astrocytes proliferate more quickly on laminin treated coverslips compared to poly-lysine treated coverslips.23 However, besides the difference in the astrocyte source compared to our study, the astrocytes were also cultured on a different material and the effect of the treatments on cell attachment was not studied in this previous paper on trout astrocytes.Collagen has been shown to increase the number of focal adhesions in cultured astrocytes under certain conditions.24 Therefore, we investigated the use of a cross-linked collagen membrane spun onto the fabricated silicon nitride membrane. Two layers of 0.3% type I collagen from bovine skin were spun on the backside of the membrane using techniques similar to spinning photoresist. Depending on the endothelial cells to be cultured, two layers of collagen were sometimes also spun on the topside of the membrane. The collagen was cross-linked with glutaraldehyde. The presence of collagen over the entire membrane backside was confirmed by positive labeling with an anti-collagen type I antibody (Fig. 5(a)), and SEM shows that the collagen structure spans the membrane pores (Fig. 5(b)). When the collagen membrane was spun on a flat wafer, the thickness, measured by SEM and profilometry, was 50 nm per dry spun layer.25
 | ||
| Fig. 5 (a) Fluorescent image showing positive labeling of collagen type I on the backside of the fabricated membrane. The entire membrane well is shown in this picture. Scale bar = 100 µm. (b) SEM picture of the spun, cross-linked collagen membrane spanning the SiN membrane pores. | ||
To test the ability of the spun collagen membranes to support astrocyte attachment in long term cultures, we grew astrocytes on solid SiN windows either spun with 2 layers of collagen or absorbed with 0.1% (1 µg cm−2) gelatin (a hydrolyzed form of collagen). The concentration for the gelatin absorption was determined from the supplier's recommendation. Each treatment was done in triplicate. The chips were removed from the medium for 30 s to simulate the TER measurement process. As shown in Fig. 6, essentially all of the astrocytes on spun, cross-linked collagen membrane remained spread and attached, whereas ∼15% of the astrocytes on gelatin absorbed membranes started to ball up and detach from the membrane. The contrast between the cells and the background is less in Fig. 6(c) compared to the other pictures shown in Fig. 6. This difference is due to the imaging set-up and does not indicate any change in the cells after air exposure that increases contrast. It was difficult to obtain more than semi-quantitative data on the percentage of cells detached from the membranes due to the astrocyte cell bodies not being distinctly clear from one another in phase contrast images. Unlike endothelial cells, astrocytes do not display contact inhibition, and will form multiple layers in cell culture. However, it is clear from Fig. 6 that the spun-on cross-linked collagen membranes support greater attachment of astrocytes in comparison to absorbed gelatin. Results similar to those for the absorbed gelatin were obtained for silicon nitride windows absorbed with 100 µg ml−1 collagen instead of gelatin (unpublished results).
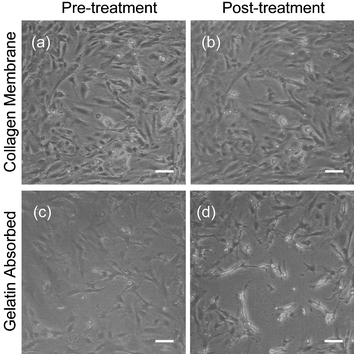 | ||
| Fig. 6 Phase contrast images of astrocytes grown on either: (a), (b) cross-linked collagen membranes on silicon nitride or (c), (d) 0.1% gelatin absorbed silicon nitride. The chips were treated by simulating the TER measurement process by removing them from the medium environment and holding them in air for 30 s. The results show that astrocytes are nearly the same before (a) and after (b) on the spun collagen membrane, but they retract and some detach (c) after treatment on the gelatin absorbed silicon nitride. The lower contrast between the cells and the background in image (c) is due to the imaging equipment and does not indicate a change in the cells upon air exposure that increases contrast. The silicon nitride windows were not patterned with pores for this experiment. Scale bar = 100 µm. | ||
Endothelial and astrocyte co-cultures on fabricated membranes
We grew several types of endothelial cells on the fabricated membranes, including BAEC, HUVEC, and bMVEC-B. For all endothelial cells except the bovine brain microvascular endothelial cells (bMVEC-B), the base/acid cleaning routine followed by adsorption with fibronectin supported cell attachment and growth for the experiment duration. However, the bMVEC-B were more sensitive to surface treatment; the commercial supplier recommended absorbing collagen and then fibronectin on cell culture membrane inserts before cell seeding. When culturing bMVEC-B on the fabricated membranes, we spun two additional layers of collagen on the topside of the chip, being careful to obtain coverage over the entire chip, and then absorbed fibronectin on top of the collagen membrane before culturing cells. This combination supported attachment and growth of bMVEC-B on the SiN membranes for the experiment duration. There were preliminary experiments in which we did not spin enough collagen to cover the entire top side of the chip surface (indicated by a dull film visible on only a portion of the top side of the chip) and the bMVEC-B grew only where the collagen had been spun. Endothelial cells and astrocytes displayed typical morphology and markers, DiI-Ac-LDL uptake and GFAP expression for endothelial cells and astrocytes respectively, when grown on the collagen spun fabricated membranes (Fig. 7). The collagen membrane on the SiN membrane also supported astrocyte attachment on the bottom side for the 12 day duration of the experiment.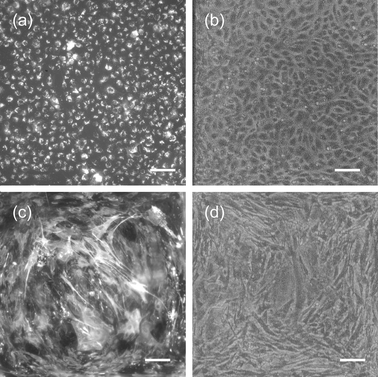 | ||
| Fig. 7 Images of endothelial and astrocyte morphology and markers on SiN membranes. (a) Fluorescent image of HUVECs showing uptake of DiI Ac-LDL . (b) Phase contrast image of HUVECs. (c) Fluorescent image of astrocytes labeled for GFAP. (d) Phase contrast image of astrocytes. Scale bar = 100 µm (a)–(d). | ||
Fig. 8 is a graph comparing TERs of the commercial and fabricated co-culture systems using commercially available bMVEC-B. For the commercial membrane portion of these experiments, Falcon PET high density membrane inserts with 0.4 µm pore size and ∼15% porosity were used. These commercial membranes are the best commercial equivalent to our fabricated membranes with 420 nm pore size and ∼25% porosity. Fig. 8 shows that resistance values for the bMVEC-B cells alone are similar for the two systems. However, using Welch's unequal variance form of the Student's t-test, these two resistances are shown to be statistically different with a significance 0.025 < p < 0.05. Assuming that the cell density on the two membranes was the same, we can explain this difference with the work of Lo et al.26 who determined that cells grown on more porous membranes should have lower TER due to a reduction in the contribution of the cell attachment to the membrane to the TER. The TER values for bMVEC-B on both the fabricated and commercial membranes were low in comparison to typical values of 200–800 Ω cm2 for good in vitro BBB models, suggesting the endothelial cells were not forming tight junctions. A negative result for the labeling of occludin (unpublished results), a membrane protein associated with tight junctions,27 supported this possibility of few tight junctions.
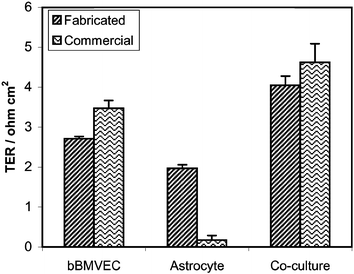 | ||
| Fig. 8 Comparison of TERs of bMVEC-B, astrocytes, and co-cultures on fabricated silicon nitride membranes with 420 nm pores and commercial membrane inserts with 0.4 µm high density pores. Measurements were taken six days post astrocyte cell seeding. | ||
Surprisingly, the TERs for the bMVEC-B alone were lower than for BAEC and HUVEC cultured on the fabricated membranes (unpublished results). When the bMVEC-B were trypsinized from the tissue culture flasks, there tended to be more cell clumps in comparison to the other endothelial cells. Before plating the endothelial cells, the cell suspension was pipetted 50+ times to break clumps of cells, but some clumps still remained. We hypothesize these clumps combined with a lower survival rate upon cell passage for the bMVEC-B compared to the BAEC and HUVEC contribute to this lower resistance value. For instance, if one of these clumps attached above the membrane in the chip well and not all of the cells in the clump survived and spread, the clump would become a low resistance area which blocks growth of higher resistance cells into that area of the membrane. Another contributing factor could be morphology differences between the cell types. HUVEC and BAEC tend to exhibit cuboidal morphology whereas bMVEC-B have a more spindly morphology as bMVEC-B tend to grow in clusters of aligned endothelial cells. Gaps where differently aligned clusters of endothelial cells grow together could be considered flaws in the cell monolayer and would tend to be low resistance areas on the membrane.28
Fig. 8 also shows there was a significant difference between the astrocyte cultures on the commercial and fabricated systems. This difference was due to a difference in astrocyte density on the two membranes. The height of the commercial membrane inserts, as well as the lack of a gasket to keep seeded cells isolated to the backside membrane area (as opposed to sticking to the plastic sides of the membrane insert or to the surrounding medium), prohibited the submersion of the entire insert in culture medium when seeding the backside with cells. Therefore, to seed cells on both sides of the commercial membranes, 45 µl of the astrocyte suspension was pipetted onto a dry membrane insert flipped upside down in a tall Petri dish. The cells were allowed to attach for 25 min, at which time the small 45 µl medium volume began to run through the membrane threatening to dry out the astrocytes. The membranes were then flipped into a 24-well plate and gravity worked to pull non-attached cells from the membrane. In contrast, the use of the silicone gasket along with submersion of both sides of the membrane in medium allowed for attachment of the astrocytes in the fabricated membrane chip well for either 8 h or overnight before the membrane was inverted. This lack of equality in attachment time probably contributed to the difference observed between the astrocyte resistances on the two membranes.
Further analysis of the data presented in Fig. 8 shows no significant difference between the co-culture resistance on the fabricated membrane compared to the sum of the endothelial and astrocyte monolayers alone, indicating two distinct cell layers were growing on opposite sides of the membrane. A direct microscopic comparison of the endothelial and astrocyte cells cultured alone versus the co-cultures on the membranes supported this separate cell layer conclusion (unpublished results). These observations support the use of a 420 nm pore size for the membranes. However, because the collagen membrane spanned the SiN membrane pores, we wanted to confirm that the collagen was not inhibiting the growth of astrocyte processes through the membranes. SEM pictures of the membrane side opposite to astrocyte seeding, similar to Fig. 4, showed small processes extending from the astrocytes in many pores (unpublished results). These processes would have the potential to directly interact with endothelial cells if they had been seeded on the top side of the membrane.
Lastly, as the co-culture resistance shown in Fig. 8 was not significantly greater than the sum of the endothelial and astrocyte cells grown alone on the fabricated membrane, no synergy in the co-culture resistance was observed. To the best of our knowledge, there have been no published TER values for these commercially available bMVEC-B co-cultured with astrocytes. However, we speculate that the lack of an increase in TER in these co-cultures is due to the primary bMVEC-B being too far removed in time from the initial cell isolation, and therefore may have lost the ability to respond to astrocyte differentiating signals. Before plating the bMVEC-B on the chip, the cells are enzymatically isolated from brain microvessels, frozen down, thawed and grown in T-flask culture in the recipient lab, and finally trypsinized and grown on the experimental membranes. For labs which do their own isolation, they are able to eliminate at least the freeze down stage, perhaps resulting in more authentic BBB endothelial cells. There has been at least one report of high resistance cultures obtained from frozen brain microvessels in co-culture with rat astrocytes when treated with cyclic adenosine monophosphate.29 The ideal test of the SiN fabricated membrane would be done with freshly isolated endothelial cells which exhibit a response to astrocyte co-culture on commercial membranes. We should also recognize that the experiment presented in Fig. 8 shows that astrocyte co-culture alone is not enough to stimulate high resistance barrier properties in endothelial cells which may not be somehow primed to respond to astrocyte signals.
There may be an optimum amount of interaction between endothelial and astrocyte cells as well as an optimum astrocyte morphology for BBB differentiation. Kacem et al.,30 have presented confocal microscopy images of rat brain slices that show astrocyte end feet form ‘rosette’-like structures (localized, interconnected, finger like branches) at the brain capillary surface. Applying imaging software (Image-Pro®) to Fig. 5(e) and (f) from Kacem et al.,30 we estimate 40% coverage of brain capillaries by astrocyte end feet. From our unpublished data, we estimate 20% coverage of the endothelial cells by astrocyte processes grown through the membrane. Furthermore, when the astrocytes are cultured alone, the processes which grow through the membrane do not have the ‘rosette’ structure observed in the intact brain capillaries. At this time, we do not have a technique developed to directly observe the morphology of the astrocyte processes which have grown through the membrane and are directly interacting with endothelial cells in our co-culture system.
There are other possible components missing from our BBB system (as well as most current in vitro BBB models). Recently other cell types, specifically neurons and pericytes, have been suggested to participate in the formation of the BBB. In a tri-culture system consisting of an immortalized rat brain capillary endothelial cell line (RBE4.B), primary rat cortical astrocytes, and primary rat cortical neurons, neurons were found to induce occludin localization at endothelial cell–cell contacts. Astrocytes led to earlier occludin localization at the cell junctions in the tri-culture system, but astrocytes were not sufficient to cause the localization when cultured alone with the endothelial cell line.31 Neurons have been found to terminate directly on BBB vessel walls.30 Pericytes are found at the abluminal BBB endothelial membrane, spaced approximately one pericyte cell for every three to four endothelial cells.32 Pericyte cell function at the BBB is not well understood. When primary rat pericytes and brain capillary endothelial cells were mixed in cell culture, tri-culture with astrocytes induced the formation of capillary-like structures from the endothelial and astrocytes.33 Lastly, in vivo, the fluids that bath the luminal (blood) and abluminal (cerebrospinal fluid) sides of the BBB capillaries have different compositions in terms of protein and ion concentrations. These fluid compositions change depending on the developmental stage of an organism.34 In our BBB model, the same fluid is used to bath both sides of the cultures; therefore our model is possibly lacking an essential polarity for BBB formation. An advantage to our model is that these different cells and media could be incorporated with little or no redesign of the system.
This membrane system has additional advantages over the traditional commercial membrane culture inserts. First, it is microscopically transparent, allowing for easy observation of cell growth on the membranes over time. When used for immunofluorescent labeling, the membrane has low background fluorescence. A further innovation of the system would be use of multiple membranes on a single chip with an adapted chamber with the ability to measure TER separately across each membrane. Multiple membranes would allow for better statistical analysis while using less valuable isolated brain microvessel endothelial cells. Similarly it could be used to measure resistances over very small areas without using such destructive methods as the device developed by Erben et al..35 There are other co-culture systems where our unique membrane properties could prove useful including fibroblast/epithelial co-cultures36 and gut epithelial/lymphocyte co-cultures.37 However, the fabricated membranes are more fragile than commercial membranes, and it is difficult to control the exact pore size due to the inherent nature of plasma etch processes without using a hard etch mask.
Conclusions
Here we demonstrated that co-culturing endothelial and astrocyte cells on membranes nanofabricated from low-stress silicon nitride is possible and is potentially useful as a unique model of the blood–brain barrier. With this model, endothelial and astrocyte co-cultures can be maintained for up to 2 weeks by using a spun-on cross-linked collagen membrane. A novel chamber to monitor and compare TERs of cells grown on the fabricated membranes has been developed. SEM pictures show that the final membrane thickness, pore size, porosity, and protein treatment allow for the potential of a high degree of direct contact between endothelial and astrocyte cells grown on opposite sides of the membrane. With the endothelial cells tested thus far, we have not observed a synergistic effect of endothelial and astrocyte co-cultures in terms of improved barrier properties. We cannot reject our original hypothesis concerning co-cultures; a more authentic endothelial–astrocyte system may respond synergistically.Acknowledgements
We would like to acknowledge Paula Miller for general cell culture assistance and Greg Baxter for helpful discussions. We thank A. Muruganadam and D. Stanimirovic of the Canadian National Research Council for the use of the SV-HCEC cell line. Financial support was provided in part by a NSF graduate fellowship and an American Association of University Women dissertation fellowship. This material is based upon work supported in part by the STC Program of the National Science Foundation under Agreement No. ECS-9876771. This work was also supported in part by a distinguished professorship awarded to Michael L. Shuler by NYSTAR (New York State Office of Science, Technology, and Academic Research). This work was performed in part at the Cornell Nano-Scale Science & Technology Facility (a member of the National Nanofabrication Users Network) which is supported by the National Science Foundation under grant ECS-9731293, its users, Cornell University and industrial affiliates.References
- U. Kniesel and H. Wolburg, Cell. Mol. Neurobiol., 2000, 20, 57 CrossRef CAS.
- H.-C. Bauer and H. Bauer, Cell. Mol. Neurobiol., 2000, 20, 13 CrossRef CAS.
- N. J. Abbott, J. Anat., 2002, 200, 629 Search PubMed.
- L. L. Rubin and J. M. Staddon, Annu. Rev. Neurosci., 1999, 22, 11 CrossRef CAS.
- A. G. de Boer, P. J. Gaillard and D. D. Breimer, Eur. J. Pharm. Sci., 1999, 8, 1 CrossRef CAS.
- M. Gumbleton and K. L. Audus, J. Pharm. Sci., 2001, 90, 1681 CrossRef CAS.
- S. Lundquist and M. Renftel, Vascul. Pharmacol., 2002, 38, 355 Search PubMed.
- C. A. Reinhardt and S. M. Gloor, Toxicol. in Vitro, 1997, 11, 513 CrossRef CAS.
- C. Lohmann, S. Huwel and H. J. Galla, J. Drug Target., 2002, 10, 263 Search PubMed.
- P. Demeuse, A. Kerkhofs, C. Struys-Ponsar, B. Knoops, C. Remacle and P. van den Bosch de Aguilar, J. Neurosci. Methods, 2002, 121, 21 CrossRef.
- Y. Hayashi, M. Nomura, S.-I. Yamagishi, S.-I. Harada, J. Yamamshita and H. Yamamoto, Glia, 1997, 19, 13 CrossRef CAS.
- I. Isobe, T. Watanabe, T. Yotsuyanagi, N. Hazemoto, K. Yamagata, T. Ueki, K. Nakanishi, K. Asai and T. Kato, Neurochem. Int., 1996, 28, 523 CrossRef CAS.
- S. G. Harris and M. L. Shuler, Biotechnology Bioprocess Eng., 2003, 8, 246 Search PubMed.
- S. Kuiper, C. J. M. van Rijn, W. Nijdam and M. C. Elwenspoek, J. Membr. Sci., 1998, 150, 1 CrossRef CAS.
- M. I. Davis-Cox, J. N. Turner, D. Szarowski and W. Shain, Microsc. Res. Tech., 1994, 29, 319 CAS.
- A. Muruganandam, L. M. Herx, R. Monette, J. P. Durkin and D. B. Stanimirovic, FASEB J., 1997, 11, 1187 Search PubMed.
- A. A. Hurwitz, J. W. Berman, W. K. Rashbaum and W. D. Lyman, Brain Res., 1993, 625, 238 CrossRef CAS.
- B. W. Kristensen, J. Noraberg, P. Thiebaud, M. Koudelka-Hep and J. Zimmer, Brain Res., 2001, 896, 1 CrossRef CAS.
- A. Sohrabi, C. Holland, R. Kue, D. Nagle, D. S. Hungerford and C. G. Frondoza, J. Biomed. Mater. Res., 2000, 50, 43 CrossRef CAS.
- C. S. Giannouilis and T. A. Desai, SPIE Micro- and Nanotechnology for Biomedical and Environmental Applications, San Jose, California, 2000, p. 122 Search PubMed.
- L. Kam, W. Shain, J. N. Turner and R. Bizios, Biomaterials, 2002, 23, 511 CrossRef CAS.
- R. Biran, M. D. Noble and P. A. Tresco, J. Biomed. Mater. Res., 1999, 46, 150 CrossRef CAS.
- E.-M. Frojdo, J. Westerlund and B. Isomaa, Comp. Biochem. Physiol. A, 2002, 133, 17 Search PubMed.
- C. Gottfried, S. R. Cechin, M. A. Gonzalez, T. S. Vaccaro and R. Rodnight, Neuroscience, 2003, 121, 553 CrossRef CAS.
- L. A. Lepak, T. Richards, N. Guillen, M. Caggana, J. N. Turner and M. G. Spencer, Mat. Res. Soc. Symp. Proc., 2003, 752, 321 CAS.
- C.-M. Lo, C. R. Keese and I. Giaever, Exp. Cell Res., 1999, 250, 576 CrossRef CAS.
- T. Hirase, J. M. Staddon, M. Saitou, Y. Ando-Adatsuka, M. Itoh, M. Furuse, K. Fujimoto, S. Tsukita and L. L. Rubin, J. Cell Sci., 1997, 110, 1603 Search PubMed.
- N. J. Abbott, C. C. W. Hughes, P. A. Revest and J. Greenwood, J. Cell Sci., 1992, 103, 23 Search PubMed.
- P. J. Gaillard, L. H. Voorwinden, J. L. Nielsen, A. Ivanov, R. Atsumi, H. Engman, C. Ringbom, A. G. de Boer and D. D. Breimer, Eur. J. Pharm. Sci., 2001, 12, 215 CrossRef CAS.
- K. Kacem, P. Lacombe, J. Seylaz and G. Bonvento, Glia, 1998, 23, 1 CrossRef CAS.
- G. Schiera, E. Bono, M. P. Raffa, A. Gallo, G. L. Pitarresi, I. Di Liegro and G. Savettieri, J. Cell. Mol. Med., 2003, 7, 165 Search PubMed.
- W. M. Pardridge, in Introduction to the Blood-Brain Barrier, New York, 1998 Search PubMed.
- M. Ramsauer, D. Krause and R. Dermietzel, FASEB J., 2002, 16, 1274 Search PubMed.
- K. M. Dziegielewska, G. W. Knott and N. R. Saunders, Cell. Mol. Neurobiol., 2000, 20, 41 CrossRef CAS.
- M. Erben, S. Decker, H. Franke and H.-J. Galla, J. Biochem. Biophys. Methods, 1995, 30, 227 CrossRef CAS.
- A. Damji, L. Weston and D. M. Brunette, Exp. Cell Res., 1996, 228, 114 CrossRef CAS.
- S. Kerneis, A. Bogdanova, J.-P. Kraehenbuhl and E. Pringault, Science, 1997, 277, 949 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |