Early mammalian embryo development depends on cumulus removal technique†
H. C.
Zeringue‡
a,
J. J.
Rutledge
b and
D. J.
Beebe
a
aDepartment of Biomedical Engineering, Department of Animal Sciences, University of Wisconsin - Madison, Madison, WI, USA
bDepartment of Animal Sciences, University of Wisconsin - Madison, Madison, WI, USA
First published on 21st July 2004
Abstract
Cumulus removal (CR) at the zygote stage is necessary for most mammalian in vitro production (IVP). Present techniques use high fluidic stresses (vortexing) or mechanical stress with enzymatic treatment (pipetting) to remove cumulus. Herein a recently developed microfluidic device for cumulus removal from zygotes is compared with traditional vortexing. Microfluidic CR (µFCR) increased development on day 2 (20 ± 4% to 35 ± 6%, p < 0.01) and blastocyst formation at day 8 (33 ± 1% to 57 ± 5%, p < 0.01) when compared to vortex CR. Vortexing effects on embryo development were studied; 15, 30 and 120 s vortex doses. Development at day 2 was inversely proportional to duration of vortexing. An in situ transcription assay was used to assess biochemical activity of zygotes after cumulus removal. There was a spike of RNA transcription of vortexed zygotes at 2 h post CR not seen in the microfluidic treatment. These results suggest the potential for microfluidic methods to enhance production efficiencies while providing insight into basic developmental mechanisms.
Introduction
Methodological advances in mammalian in vitro embryo production (IVP) have increased the scope and application of assisted reproduction in several species. Intracytoplasmic sperm injection1 (ICSI) and cloning by nuclear transfer2 (NT) and nuclear microinjection3 (MI) have increasingly been used in research and clinical settings4 and benefit from robust oocytes and embryos.5 Microtechnologies have also found application in assisted reproduction and have begun to engage IVP,6,7 male gamete manipulation8 and cellular biology.9 Cumulus removal (CR) at the zygote stage is necessary for much of mammalian IVP. Present techniques (vortexing and pipetting) use high stresses and/or enzymatic treatment to remove cumulus. We have recently developed a microfluidic cumulus removal (µFCR) device for mammalian zygotes.10 We present data showing that µFCR enhances early embryo development relative to vortexing and that an initial developmental latency due to vortexing is dose dependant. In addition, an in situ assay confirms that the two CR techniques cause fundamental differences in the dynamics of early embryo RNA transcription. These data suggest that assisted reproduction (ICSI, NT, MI) may benefit from µFCR.Several layers of cumulus cells surround the oocyte when it is retrieved from a donor. Cumulus cells aid in oocyte maturation11 and fertilization,12,13 but must be removed after these events. There are two common methods, developed decades ago, for in vitro cumulus removal; flushing14 and vortexing.15 Flushing involves repeated, vigorous aspiration and ejection of the zygote through a pipette having an internal diameter slightly smaller than the zygote, often in the presence of enzymes.16,17 Vortexing involves placing a group of zygotes in a vial with a small amount of fluid and then agitating with a laboratory vortex for several minutes. Vortexing is widely used when large numbers of zygotes must be processed and has the advantage of eliminating the opportunity for enzymatic damage.15
Experimental procedure
For all experiments, oocyte collection, maturation and fertilization were done as previously described.18 Cattle oocytes were aspirated from abattoir ovaries and placed in maturation medium for 24 h at 39 °C, 5% CO2 in air. Oocytes were then co-incubated in fertilization medium with sperm for 24 h at 39 °C, 5% CO2 in air. At 24 h post fertilization, zygotes were distributed among treatment groups and cumulus was removed as below. Zygotes were placed in 50 µL culture medium drops (20–24 zygotes per drop) for culture at 39 °C, 5% CO2 in air. Culture medium was SOF (Specialty Media, Phillipsburg, NJ) supplemented with 0.4 mM sodium pyruvate, 8 mg mL−1 fatty acid-free BSA nonessential amino acid solutions (Sigma) and 25 µg mL−1 gentamycin. Heat-treated fetal bovine serum (Gibco BRL, Grand Island, NY) was added to each drop five days after fertilization to give a final concentration of 10% (v/v).Microfluidic vs. vortex cumulus removal
Experiment 1 compared µFCR and vortex cumulus removal. Three treatment groups were used; (A) µFCR experimental group, (B) vortex control group briefly (1–3 min) placed in a microfluidic environment and (C) a vortex control group. At 24 h post-fertilization (PF), zygotes were randomly distributed among treatment groups and cumulus was removed. Group A zygotes were placed in warmed (39 °C) microfluidic devices pre-filled with TL-HEPES (BioWhittaker, Walkersville, MD; supplemented with 0.2 mM sodium pyruvate and 50 µg mL−1 gentamycin throughout all experiments). One to three zygotes were placed in the device at a time. Cumulus was stripped using hand-modulated syringe pressure (see Electronic supplementary information (ESI†). Group A zygotes were retrieved from the device after cumulus removal (typically 1–3 min), washed in TL-HEPES, and placed into culture. Group B and C zygotes were placed in a 1.5 mL microcentrifuge tube containing 400 µL TL-HEPES. The tube was held in a laboratory vortex at high for 3 min. Group B zygotes were removed from the tube and placed in a microfluidic device filled with TL-HEPES for 3 min, retrieved, washed, and placed into culture. Group C zygotes were washed and placed directly into culture following vortexing. Visual observation assured complete cumulus removal from each method (results not shown). Developmental recordings were taken at 48 h PF (Day 2) and 192 h PF (Day 8). There were nine replicates with approximately 136 cleaved embryos in each treatment group. Total zygotes processed; group A = 208, group B = 206, group C = 210. Total embryos cleaved; group A = 134, group B = 136, group C = 139.Statistical analysis was a paired Student's t employed orthogonally with one comparison between controls (B vs. C) and the second comparison between control groups average and experimental group (avg[B,C] vs. A).
Vortex dose response
In experiment 2, oocytes were collected, matured and fertilized as above. At 24 h PF, zygotes were randomly distributed among three different treatment groups. Vortexing was done for 15 s (Group 15), 30 s (Group 30) or 120 s (Group 120) at 24 h PF. When necessary, remaining cumulus was removed using µFCR to ensure cumulus was completely removed from all treatment groups. Zygotes were then placed in culture as above. Developmental readings were taken at 48 h PF (Day 2). Each treatment group contains approximately 33 cleaved embryos. Statistical analysis of mean cell count was by one-way analysis of variance (ANOVA). There were 32–34 replicates. Analysis was robust as tested with the method described by Box.19In situ transcription assay
Zygotes were randomly distributed among treatment groups and cumulus was removed by either µFCR or vortex method. Nude zygotes were then placed into culture. Transcription assay was performed at either 2 h, 8 h or 24 h as previously described.20 Briefly, the zygote was washed in protein-free buffer then the cell membrane was permeabilized in 0.05% Triton-X (Sigma) for 1–2 min, then incubated with bromouridine triphosphate (BrUTP) (Sigma) for 15 min. After nuclear membrane permeabilization in 0.2% Triton-Z for 3 min, zygotes were fixed in 3.7% paraformaldehyde. Zygotes were incubated with anti-BrdU mouse IgG1 monoclonal antibody (Molecular Probes, Eugene, OR) then fluorescent goat anti-mouse IgG1 (Molecular Probes) to fluorescently label BrUTP incorporated RNA. Statistical analysis of average image intensity was by two-way ANOVA with the F value comparing the mean square interaction (MSint) to the mean square within (MSw).Results
Herein a recently developed microfluidic device10 for cumulus removal from zygotes is compared with traditional vortexing. Microfluidic CR increased development on day 2 (20 ± 4% to 35 ± 6%, p < 0.01) (as judged by percent having undergone 3 cleavages) and blastocyst formation at day 8 (33 ± 1% to 57 ± 5%, p < 0.01) when compared to vortex CR. Vortexing effects on embryo development were studied using a dose response experiment (15, 30 and 120 s). Development at day 2 was inversely proportional to duration of vortexing. An in situ transcription assay was used to assess biochemical activity of zygotes after cumulus removal. Vortexed zygotes had a spike of RNA transcription 2 h post cumulus removal (pCR) not seen in the microfluidic treatment. These results suggest the potential for microfluidic methods to enhance production efficiencies while providing insight into basic developmental mechanisms.The µFCR device uses syringe controlled fluid flow to both manipulate the zygote and strip any cumulus cells. Cumulus is drawn into a narrow removal port (Fig. 1(a) arrow). Hand-modulated pulses in fluid flow allow zygote rotation and removal of all cumulus cells. After cumulus has been removed through the port, the denuded zygote is retained in the channel for retrieval (Fig. 1(b)). Experiment 1 compares µFCR with vortex cumulus removal. Experiment 2 investigates the dependence of early embryo development on duration of vortexing. Experiment 3 uses an in situ transcription assay to explore metabolic differences between µFCR and vortex CR zygotes as assessed by newly formulated RNA.
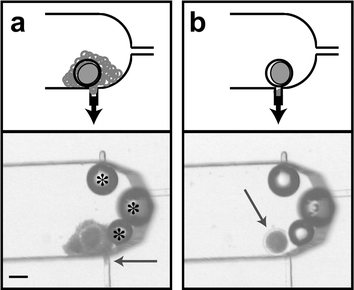 | ||
| Fig. 1 Cumulus removal of zygotes in a microchannel (ESI†). A schematic representation is shown above each photomicrograph. (a) The cumulus is seen being drawn into the removal port (arrow). Bubbles (*) are stable in the device and do not affect cumulus removal. (b) After cumulus removal, the cleaned zygote (arrow) is ready to be retrieved from the channel for further in vitro processing. Scale bar is 100 µm. | ||
Experiment 1 developmental data (Fig. 2) is represented as a percentage of the total number of cleaved embryos on day 2 (48 h post fertilization (PF)). There were no differences (p > 0.6) in the number of embryos that cleaved at least once between the three treatment groups. There was no difference between control groups (Groups B and C) at any time point. On day 2, there were more (p < 0.01) 8-cell stage embryos in the µFCR group than the control groups (35 ± 6% and 20 ± 4%, respectively). On day 8, there were more blastocysts (57 ± 5% compared to 33 ± 1%, p < 0.01) and expanded blastocysts (39 ± 6% compared to 17 ± 1%, p < 0.01) in the µFCR group than in the control groups.
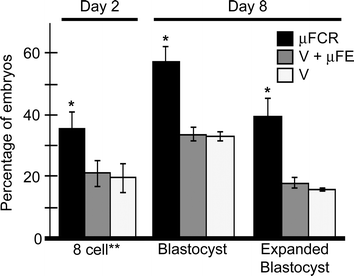 | ||
| Fig. 2 Developmental data from cumulus removal study. Day 2 data is the percentage of 8 cell embryos of total number cleaved in each treatment. Day 8 data is the percentage of blastocyst and expanded blastocyst. Treatment A (black, n = 134) underwent microfluidic cumulus removal (µFCR). Treatment B (grey, n = 136) underwent vortex (V) cumulus removal followed by placement in a microfluidic environment.33 Treatment C (white, n = 139) underwent vortex cumulus removal. (*) Represents a significant difference (p < 0.01) from the other columns. (**) Embryos with 6 or more cells at the time of reading were categorized as 8-cell. | ||
For experiment 2, embryos were categorized into 2-, 4- or 8-cell stages at day 2. Embryo development data shown in Fig. 3 are a percentage of total cleaved embryos (group 15 (n = 34), group 30 (n = 32), group 120 (n = 34)). As the length of vortexing increases (from 15 to 120 s), the percentage of 8-cell embryos decreases (18% to 0%), while the percentage of 2 cell embryos increases (24% to 39%). The percentage of 4-cell embryos remains relatively constant (∼59%) across treatment groups. The data shows a clear decrease in the number of cells per embryo at 48 h PF resulting from increased vortex duration at 24 h PF (F(2,98) = 3.38, p < 0.05).
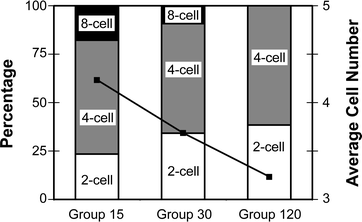 | ||
| Fig. 3 Embryo development at Day 2 for vortex dose experiment. Embryos were vortexed for 15 s (Group 15), 30 s (Group 30) or 120 s (Group 120). Vertical bars represent percentage of embryos by stage (primary axis). Black squares indicate the average cell number per embryo (secondary axis). There is a significant decrease in cell count with increased vortexing time (F(2,98) = 3.4, p < 0.05). The pooled within cell mean square was 2.52. All values are out of cleaved embryos; n group15 = 34, n group30 = 32, n group120 = 34. (**) Embryos with 6 or more cells were categorized as 8-cell. | ||
Experiment 3 incorporates a tagged ribonucleic acid (bromouridine (BrU)) into transcribed RNA for 15 min. The fluorescence intensity is proportional to transcription activity. The experiment explores the transcription activity between µFCR and vortexed zygotes at 2 h, 8 h and 24 h pCR. Fig. 4(a) shows fluorescent images of a zygote from each treatment group. Fig. 4(b) charts the average fluorescence intensity from images of zygotes in each treatment group (for each group n = 4). Transcription activity of vortexed zygotes was initially high and decreased during the first 24 h pCR. Microfluidic CR zygotes presented an opposite trend in transcription activity (F(2,18) = 12.08, p < 0.001).
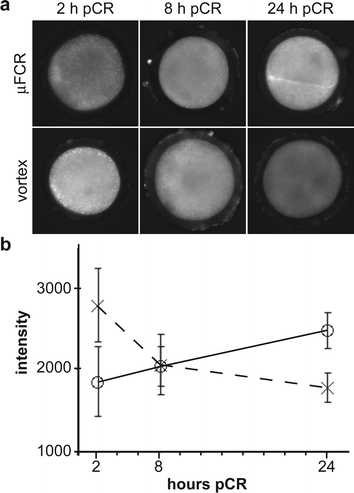 | ||
| Fig. 4 Transcription activity of zygotes following vortexing and µFCR. (a) Photomicrographs of fluorescently labeled zygotes. Bromouridine (BrU) incorporated during 15 min RNA transcription at 2, 8 and 24 h post cumulus removal (pCR). Brighter fluorescence indicates higher levels of RNA transcription. (b) Graph of average fluorescent intensity vs. time for µFCR (○) and vortexed (×) zygotes. Fluorescence intensity increases with time in the µFCR zygotes (solid line), but decreases in vortexed zygotes (dotted line). | ||
Discussion
Experiment 1 compares the µFCR and vortex CR techniques. Group A represents the experimental microfluidic approach. Group B accounts for the effect of the microfluidic environment on development when the control CR (e.g. vortex) is employed. Group C represents the control CR approach. At each recording, there was no difference between Groups B and C (Fig. 2), suggesting that the brief (1–3 min) exposure to a microfluidic environment at 24 h PF has no effect on early embryogenesis.Day 2 data from experiment 1 shows a decrease in 8 cell stage embryos when vortexing is employed. Forces associated with vortexing and mixing are known to cause damage and death21 as well as changes in metabolic function22 in other mammalian cells. Vortexing creates large shear stresses and acceleration forces in the fluid. Large forces are indirectly caused as vortex induced bubbles collapse at the air/liquid interface. When these forces are removed from the processing protocol by replacing vortexing with µFCR, embryos are shown to fare better. These data are consistent with the hypothesis that vortexing causes developmental injury to early stage embryos.
Experiment 2 shows the utility of microfluidics to reveal small changes in development rates. Usually, cumulus is incompletely removed when zygotes are vortexed for less than 2 min. Any cumulus remaining on zygotes after these short vortex doses is removed with the microfluidic device. If excess cumulus were left on the zygotes during culture, one would not be able to differentiate the developmental changes brought about by differing amounts of cumulus from changes due to vortex duration. The µFCR technique allows excess cumulus to be removed without causing undue developmental stress to the embryo (results of experiment 1). Experiment 2 exposed embryos to three different vortex durations (15 s, 30 s, and 120 s). There is a shift towards lower cell counts as vortex duration increased (Fig. 3). Because cleavage rate is equivalent in each group (∼75%), data does not suggest vortex-induced fatal injury during period studied. The data does suggest a decrease in developmental competency that is proportional to the duration of vortexing. It should be noted that many protocols suggest vortexing for at least 2 min to ensure complete cumulus removal.23–25
Experiment 3 reveals differences in transcription activity between zygotes processed using µFCR and vortex CR methods. When compared with µFCR, vortexing causes a spike in transcription activity during the first 2 h following CR. This spike pattern of transcription activity between µFCR and vortex CR is similar to the transcription activity patterns reported from in vivo-derived and in vitro-cultured mouse embryos.26 In the mouse, a spike of heat-shock protein 70 (HSP70, a repair protein) was seen at the 2-cell stage of in vitro-cultured embryos when compared with in vivo-derived embryos. Production of repair proteins is consistent with the notion of vortex-induced damage.
These results demonstrate effects of present processing techniques on embryos and benefits of adapting microfluidic techniques to embryology. In the first two experiments, microfluidic handling significantly increases early embryo developmental rates. The rate of embryo development (i.e. from fertilization through blastocyst formation) is often referred to as ″developmental kinetics.″ Some researchers report that increased developmental kinetics may not benefit, and may actually decrease, embryo viability.27 We suggest our increased development is not due to an artificial increase in kinetics. Rather, removing the stresses incurred during vortexing allows embryo development to proceed unimpeded. Microfluidic handling yields increased development, suggesting a less traumatic in vitro process. This gentler µFCR process allowed for a more sensitive system in which to test embryos. Experiment 2 demonstrates the ability of microfluidic processing to help differentiate developmental differences occurring from small perturbations in embryo processing. Experiment 3 shows that microfluidic techniques can fundamentally change embryogenesis at the molecular level leading to potential production implications such as large offspring syndrome in cattle.28
These results pave the way towards validating the utility of microfluidics in assisted reproduction. Understanding the cellular and molecular basis of in vivo embryogenesis will facilitate the assessment of in vitro procedures in producing in vivo-like embryos. Researchers are attempting to gain a greater understanding of the mechanisms underlying mammalian embryogenesis.29–32 Until more is understood about the basic biology surrounding embryogenesis,4,29 the origin of these observations will remain unknown. We believe the combination of microfluidics and embryology will not only allow for more embryo-friendly processes, but also facilitate greater understanding of the basic science surrounding embryogenesis.
Acknowledgements
The authors thank Rick Monson (UW-Madison) and Matt Wheeler (University of Illinois at Urbana-Champaign) for helpful discussions and technical assistance. H.Z. thanks F. Aoki (University of Tokyo) for help in establishing the BrUTP protocol. This research was funded in part by a UW UIR-IEDR grant and NLM 5T15LM005359 (H.Z.).References
- G. Palermo, H. Joris, P. Devroey and A. C. V. Steirteghem, Pregnancies after intracytoplasmic sperm injection of single spermatozoon into an oocyte, Lancet, 1992, 340, 17–18 CrossRef CAS.
- I. Wilmut, A. E. Schnieke, J. McWhir, A. J. Kind and K. H. Campbell, Viable offspring derived from fetal and adult mammalian cells, Nature, 1997, 385, 810–813 CrossRef CAS.
- T. Wakayama, A. C. Perry, M. Zuccotti, K. R. Johnson and R. Yanagimachi, Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei, Nature, 1998, 394, 369–374 CrossRef CAS.
- R. M. Schultz and C. J. Williams, The science of art, Science, 2002, 296, 2188–2190 CrossRef CAS.
- S. Kattera, P. Shrivastav and I. Craft, Comparison of pregnancy outcome of pronuclear- and multicellular-stage frozen-thawed embryo transfers, J. Assist. Reprod. Genetics, 1999, 16, 358–362 Search PubMed.
- D. J. Beebe, M. Wheeler, H. C. Zeringue, E. Walters and S. Raty, Microfluidic technology for assisted reproduction, Theriogenology, 2002, 57, 125–135 CrossRef CAS.
- S. Raty, J. A. Davis, D. J. Beebe, S. L. Rodriguez-Zas and M. B. Wheeler, Culture in microchannels enhance in vitro embryonic development of preimplantation mouse embryos, Theriogenology, 2001, 55, 241.
- B. S. Cho, T. G. Schuster, X. Y. Zhu, D. Chang, G. D. Smith and S. Takayama, Passively driven integrated microfluidic system for separation of motile sperm, Anal. Chem., 2003, 75, 1671–1675 CrossRef CAS.
- A. Sawano, S. Takayama, M. Matsuda and A. Miyawaki, Lateral propagation of egf signalling after local stimulation is dependent on receptor density, Developmental Cell, 2002, 3, 245–257 Search PubMed.
- H. C. Zeringue, D. J. Beebe and M. B. Wheeler, Removal of cumulus from mammalian zygotes using microfluidic techniques, Biomed. Microdevices, 2001, 3, 219–224 CrossRef.
- Y. Fukui and Y. Sakuma, Maturation of bovine oocytes cultured in vitro: Relation to ovarian activity, follicular size and the presence or absence of cumulus cells, Biol. Reprod., 1980, 22, 669–673 Search PubMed.
- R. C. Chian, K. Okuda and K. Niwa, Influence of cumulus cells on in vitro fertilization of bovine oocytes derived from in vitro maturation, Anim. Reprod. Sci., 1995, 38, 37–48 CrossRef.
- A. Martino, T. Mogas, M. J. Palomo and M. T. Paramio, In vitro maturation and fertilization of prepubertal goat oocytes, Theriogenology, 1995, 43, 472–485.
- D. P. Wolf, in In vitro Fertilization and Embryo Transfer, ed. D.P. Wolf, Plenum Press, New York, 1988, 132––135 Search PubMed.
- A. G. Sacco and C. A. Shivers, in Methods in Mammalian Reproduction, ed. J.C. Daniel, Academic Press, New York, 1978, 214––216 Search PubMed.
- A. S. Doherty and R. M. Schultz, in Developmental Biology Protocols, ed. R. S. Tuan and C.W. Lo, Humana Press, Totowa, NJ, 2000, vol. 1, pp. 50–51 Search PubMed.
- B. Hogan, R. Beddington, F. Costantini and E. Lacy, Manipulating the Mouse Embryo, Cold Spring Harbor Laboratory Press, Cold Harbor Springs, NY, 2nd edn., 1994 Search PubMed.
- A. Fisher, D. P. Bernal, C. Guiterrez-Robayo and J. J. Rutledge, Estimates of heterosis for in vitro embryo production using reciprocal crosses in cattle, Theriogenology, 2000, 54, 1433–1442 CrossRef.
- G. E. P. Box, Some theorems on quadratic forms applied in the study of analysis of variance problems, i. Effects of inequality of variance in the one-way classification, Ann. Math. Stat., 1954, 25, 290–302 Search PubMed.
- F. Aoki, D. M. Worrad and R. M. Schultz, Regulation of transcriptional activity during the first and second cell cycles in the preimplantation mouse embryo, Develop. Biol., 1997, 181, 296–307 Search PubMed.
- J. Wu, Mechanisms of animal cell damage associated with gas bubbles and cell protection by medium additives, J. Biotechnol., 1995, 43, 81–94 CrossRef CAS.
- S. Mercille, M. Johnson, S. Lanthier, A. A. Kamen and B. Massie, Understanding factors that limit the productivity of suspension-based perfusion cultures operated at high medium renewal rates, Biotechnol. Bioeng., 2000, 67, 465–450 CrossRef.
- P. J. M. Hendriksen, W. N. M. Steenweg, J. C. Harkeme, J. S. Merton, M. M. Bevers, P. L. A. M. Vos and S. J. Dieleman, Effects of different stages of the follicular wave on in vitro developmental competence of bovine oocytes, Theriogenology, 2004, 61, 909–920 CrossRef CAS.
- G. Vajta, T. T. Peura, P. Holm, A. Paldi, T. Greve, A. O. Trounson and H. Callesen, New method for culture of zona-included or zona-free embryos: The well of the well (wow) system, Mol. Reprod. Develop., 2000, 55, 256–264 Search PubMed.
- P. Lonergan, D. Rizos, A. Gutierrez-Adan, P. M. Moreira, B. Pintado and J. de la Fuente, Temporal divergence in the pattern of messenger rna expression in bovine embryos cultured from zygotes to blastocyst stage in vitro or in vivo, Biol. Reprod., 2003, 69, 1424–1431 Search PubMed.
- E. Christians, E. Campion, E. M. Thompson and J. P. Renard, Expression of the hsp 70.1 gene, a landmark of early zygotic activity in the mouse embryo, is restricted to the first burst of transcription, Development, 1995, 121, 113–122 Search PubMed.
- M. Lane and D. K. Gardner, Differential regulation of mouse embryo development and viability by amino acids, J. Reprod. Fertility, 1997, 109, 153–164 Search PubMed.
- G. Lazzari, C. Wrenzycki, D. Herrmann, R. Duchi, T. Kruip, H. Niemann and C. Galli, Cellular and molecular deviations in bovine in vitro-produced embryos are related to the large offspring syndrome, Biol. Reprod., 2002, 67, 767–775 Search PubMed.
- J. Eckert and H. Niemann, Mrna expression of leukaemia inhibitory factor (lif) and its receptor subunits glycoprotein 130 and lif-receptor-beta in bovine embryos derived in vitro or in vivo, Mol. Hum. Reprod., 1998, 4, 957–965 CrossRef CAS.
- P. Hyttel, et al., Ribosomal rna gene expression and chromosome aberrations in bovine oocytes and preimplantation embryos, Reproduction, 2001, 122, 21–30 Search PubMed.
- R. W. Wright and J. Ellington, Morphological and physiological differences between in vivo- and in vitro-produced preimplantation embryos from livestock species, Theriogenology, 1995, 44, 1167–1189 CrossRef.
- H. Niemann and C. Wrenzycki, Alterations of expression of developmentally important genes in preimplantation bovine embryos by in vitro culture conditions: Implementations for subsequent development, Theriogenology, 2000, 53, 21–34 CrossRef CAS.
- G. M. Walker, H. C. Zeringue and D. J. Beebe, Microenvironment design considerations for cellular scale studies, Lab Chip, 2004, 4, 91–97 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Microfluidic cumulus removal from a bovine zygote. See http://www.rsc.org/suppdata/lc/b3/b316494m/ |
| ‡ Present address: Department of Biology 68-380, Massachusetts Institute of Technology, Cambridge, MA, USA. |
| This journal is © The Royal Society of Chemistry 2005 |