Photochemistry of 6-chloro and 6-bromopicolinic acids in water. Heterolytic vs. homolytic photodehalogenation
Florence
Rollet
,
Claire
Richard
* and
Jean-François
Pilichowski
Laboratoire de Photochimie Moléculaire et Macromoléculaire, UMR CNRS-Université Blaise Pascal 6505, 63177, Aubière Cedex, France. E-mail: claire.richard@univ-bpclermont.fr; Fax: +33 4 73 40 77 00; Tel: +33 4 73 40 71 42
First published on 18th October 2005
Abstract
The photochemistry of 6-chloro and 6-bromopicolinate ions (I and II, respectively) was investigated by product studies and ns laser flash photolysis (LFP). In deoxygenated pH 5.4 water, I yields 6-hydroxypicolinic acid (70%) and a substituted pyrrole. In 2-propanol–water (1 : 1) mixture, the reaction yields, very unselectively, 6-hydroxypicolinic acid, 2-carboxypyridine, pyridine and bipyridines. Photolysis of aqueous II leads to 6-hydroxypicolinic acid (78%) and hydroxybipyridines. Oxygen suppresses the photolysis of I but does not affect that of II. By LFP, we detected a short-lived transient at the pulse end from I (λmax = 305 nm, k = (2.8 ± 0.2) × 105 s−1, εϕ = 2200 ± 200 dm3 mol−1 cm−1). This is quenched either by oxygen or methyl acrylate and thus assigned to the triplet excited state. The triplet excited state of II is detected at pH 1 only (λmax = 320 nm, k > 3 × 107 s−1). The radical ion Cl2−˙ could be successfully detected by photolysing I in 2-propanol–water (1 : 1) in the presence of Cl−. Similarly, Br2−˙ could be detected by irradiating aqueous II in the presence of Br−. These results show that the photodehalogenation of I is heterolytic in water and mainly homolytic in 2-propanol–water mixtures while that of II is both heterolytic and homolytic in water. A mechanism in which the triplet excited state undergoes homolysis of the C–X bond and subsequent electron transfer from the carboxypyridyl radical to the halogen atom to form an ion pair may account for these observations.
1 Introduction
The photochemistry of chloro–aryl compounds has been extensively studied because these compounds are environmental pollutants. The reactions involved are complex. Irradiation of chloro–aryls in solution affords dehalogenation, but the nature of the photoproducts is highly solvent dependent. In non-polar solvents, reduction products and biphenyls are formed. They arise from homolytic cleavage of the C–Cl bond followed either by the abstraction of a H atom from the solvent by the aryl radical or by the addition of it to a molecule of the starting compound.1–7In pure water or mixed water–organic solvents, photosubstitution of the halogen by an OH group competes with photoreduction.8–10 It is often the major pathway. Unlike photoreduction, photohydrolysis is explained by a heterolytic cleavage of the C–Cl bond. To rationalize this dual behaviour, it was proposed that radicals are initially produced by homolytic cleavage of the C–Cl bond. Then, they either escape the cage or undergo electron transfer to afford an ion pair. This mechanism was first demonstrated in the case of alkyl halides.11
Halogen substituted nitrogen heterocycles are also an important class of molecules from a biological point of view, but they have been much less studied than halogeno–aryls. Like these latter, they show ionic and radical photodehalogenation in polar solvents.12–16 Hence, 2-chloropyrimidine was recently found to undergo both hydrolysis and dimerization with a low quantum yield (≈0.015) when irradiated in water.16 The selective inhibiting effect of oxygen on dimer formation, the D2O/H2O isotopic effect on photohydrolysis (1.4) and the sensitization of the reaction by acetone prompted the authors to propose that hydrolysis took place via the singlet excited state and dimerization via the triplet.
We decided to investigate the photochemical behaviour of other halogen substituted nitrogen heterocycles in order to get a better insight in the mechanism of dehalogenation. In a previous work, we studied the photochemistry of aqueous 2, 3 and 4-carboxypyridines in detail17 and found that the ortho isomer was the most photoreactive. For this reason and as an extension of this former work, we have studied the photochemistry of two halogen substituted compounds: 6-chloro- and 6-bromo-2-carboxypyridine (I) and (II) (see Scheme 1).
 | ||
| Scheme 1 | ||
2 Experimental
2.1 Materials
6-Chloro-2-carboxypyridine (I), 6-bromo-2-carboxypyridine (II), 6-hydroxy-2-carboxypyridine (III) and 2-carboxypyridine were purchased from Aldrich (99% purity grade) and used as received. All other reactants were of the highest grade available. Water was purified using a Millipore (Milli-Q) device.2.2 Steady state irradiations
For quantitative purposes, aqueous solutions of I or II were irradiated at 280 nm in a quartz cuvette (1 cm optical length) using an arc xenon lamp (1600 W) equipped with a Schoeffel monochromator. The bandwidth to middle height was 10 nm. Solutions were deoxygenated by argon bubbling or oxygenated by oxygen bubbling for 20 minutes prior to irradiation. Then the cuvette was closed using a septum. For analytical purposes, irradiations were performed in a device equipped with six germicide lamps emitting at 253.7 nm and a 100 ml cylindrical quartz reactor. Potassium ferrioxalate was used as a chemical actinometer. The pH of the solutions was adjusted using phosphate buffers (10−3 M, pH = 5.4) or with HClO4.2.3 Laser flash photolysis
Laser flash photolysis experiments were carried out using an Applied Photophysics LKS.60 apparatus equipped with a Nd3+:YAG laser Quanta-Ray GCR-130. The experimental procedure was described elsewhere.18 Samples were irradiated using the fourth harmonic (266 nm, pulse duration 9 ns) in a quartz cuvette. Solutions were deoxygenated by nitrogen bubbling directly in the cell and changed after each shot to avoid excessive exposure. Potassium peroxodisulfate was used as a chemical actinometer. The εϕ product for the formation of SO4−˙ at 450 nm was taken as 1880 dm3 mol−1 cm−1.19 The solutions of the actinometer and of the substrates were diluted in order to show the same absorbance at 266 nm. The εϕ products were estimated using the linear variations of absorbance changes with laser pulse energy.2.4 Analyses
UV-visible spectra were recorded on a Cary 3 (Varian) spectrophotometer. Proton and carbon NMR spectra were collected on 300 and 500 Avance Bruker spectrometers, depending on the program used. The products were analyzed, at 23 °C, in solution in D2O (4.8 ppm), CDCl3 (7.28 ppm) or DMSO-d6 (2.59 ppm) and the solvent was used as an internal reference. HPLC-ESI-MS spectrometry analyses were performed by the Service Central d'Analyse of CNRS (Solaize, France). A Hewlett-Packard model HP 1100-MSD was used to obtain the mass spectra. Products were separated on a column Waters X-Terra MS 3.5 µm C18 (2.1 mm × 150 mm). The temperature of the column was maintained at 50 °C. A gradient of elution: HCO2H-acidified water (pH = 2.6)–acetonitrile was used as eluent at a flow of 0.3 ml min−1. Nitrogen was used as sheath gas at 13 L min−1 and the spray voltage was 3 kV. Consumption of starting compounds and formation of photoproducts were monitored by analytical HPLC using a Waters apparatus equipped with a 996-photodiode array detector and a column Varian Omnisphere 5 µm C18 (4.6 mm × 250 mm). The mobile phase was a mixture of H3PO4-acidified water (pH = 2.7)–methanol (70/30%, v/v). The pH meter was a 3310 Jenway. It was equipped with an Ag/AgCl glass combination electrode 9102 Orion. The formation of chloride ions in solution was monitored by ionic chromatography using an apparatus equipped with a detector of conductivity of the type Waters 431, a pump of the type Gilson 305, a degasser G1379A Agilent series 1100 and an injector connected to an anion column of type IC-Pak (4.6 mm × 150 mm). The mobile phase was a mixture of borate–gluconate (glycerin + sodium gluconate + boric acid + sodium tetraborate). This method required us to plot a calibration curve using solutions of known Cl− concentration. A deoxygenated solution of I (9.3 × 10−5 mol dm−3, pH 4.2) was irradiated at 280 nm for 75 minutes to achieve a conversion extent of 20%. This sample was then analyzed by ionic chromatography with a flow of 2 ml min−1 and on a sensitivity regulated to 0.001.2.5 Preparative-scale irradiations for photoproducts identification
3 Results and discussion
3.1 Spectral characteristics
The absorption spectra of I and II in pH 5.4 buffered water show maxima at 275 ± 2 nm with εmax equal to 4700 ± 100 and 4300 ± 100 dm3 mol−1 cm−1, respectively. They are red-shifted in comparison to that of 2-carboxypyridine, the non-halogenated counterpart (λmax = 263 nm, εmax = 4100 ± 100 dm3 mol−1 cm−1). I and II show two pKas just as 2-carboxypyridine. Using spectrophotometry, we could estimate that the values of pKa1 were very low (<0.5). The values of pKa2 are reported in the literature. These are equal to 3.27 and 3.25, respectively,20 against 5.29 for 2-carboxypyridine.21I and II are thus much stronger acids than 2-carboxypyridine due to the presence of halogens that are electron-withdrawing substituents. Most of the experiments were performed in pH 5.4 buffered water so that I and II were almost entirely in the anionic form.3.2 Steady-state irradiation of I
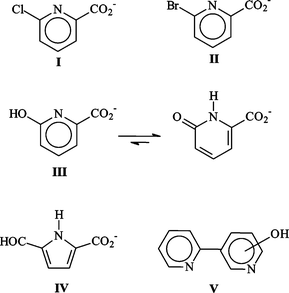
As shown in Fig. 1, irradiation of deoxygenated aqueous I yielded two organic photoproducts, cleanly. The reaction was found to be independent of the initial substrate concentration within the range 10−4–10−3 mol dm−3. The first eluted photoproduct (λmax = 308 nm, see Fig. 1) was assigned to 6-hydroxy-2-carboxypyridine (III) by comparison with an authentic sample. We note that this compound is mainly present in the ketonic form in water. To allow the identification of the second photoproduct (IV), we irradiated a concentrated solution of I until a high conversion extent (≈ 90%) was achieved, after having checked that the unknown photoproduct was not photolysed in turn. We also verified that the ratio of the photoproducts in the mixture remained constant over the whole reaction. NMR analysis of the crude residue in DMSO revealed the presence of two non-D2O-exchangeable doublets (δ = 6.50 and 6.85 ppm with J = 4 Hz), of a non-D2O-exchangeable singlet at δ = 9.6 ppm and of a D2O-exchangeable broad resonance at 13 ppm along with resonances related to III and the remaining I (Fig. 2). The doublets can be assigned to coupled cis-vinylic protons, the non-exchangeable singlet to an aldehyde and the broad exchangeable resonance to a carboxylic proton. The molecular weight of IV (139) corresponded to the stoichiometry I–HCl + H2O. These data as well as the presence of an aldehydic proton and the strong and broad absorption band with maximum at 298 nm (see Fig. 1) claim in favour of a pyrrole structure bearing CO2H and CHO substituents in positions 2 and 5 (see Scheme 1).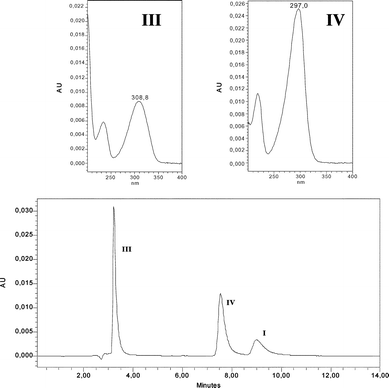 | ||
| Fig. 1 Bottom: HPLC chromatogram of irradiated deoxygenated water solution of anionic I (10−4 mol dm−3). Conversion extent of 88%. Top: UV spectra of III and IV. | ||
 | ||
| Fig. 2 Proton NMR spectrum of an irradiated solution of I. Conversion extent of 20%. A few drops of D2O were added to the initial solution in DMSO-d6. | ||
Quantum yields of I photolysis and of photoproducts formation were measured upon irradiation at 280 nm. Data are reported in Table 1. In deoxygenated pH 5.4 water, the quantum yield of I photolysis (ϕClPa) was equal to 0.062. To titrate chloride ions using ionic chromatography, we had to photolyse I in pure water. In these pH conditions (pH ≈ 4.2), ϕClPa and the quantum yield of Cl− formation were found to be both equal to 0.038, indicating that the chlorine atom is released by a heterolytic process only. In buffered and unbuffered solutions, II accounted for 70% of converted I. In order to investigate the influence of solvent polarity on the reaction, we photolysed I in a 1 : 1 2-propanol–pH 5.4 water mixture. In these conditions numerous photoproducts were detected. We firmly identified III, 2-carboxypyridine and pyridine. The other compounds showed absorption spectra and retention times that resemble those of substituted bipyridines found in a previous work.17 Addition of 2-propanol (50% in volume) significantly increased ϕClPa that reached 0.16, but drastically decreased the quantum yield of III formation as well as that of IV. Clearly, addition of 2-propanol strongly affected the reaction favouring free radical chemistry. The effect of solution oxygenation on the reactions was also studied. Photolysis of I was drastically inhibited by oxygen, whatever the solvent used.
| Quantum yield | pH 4.2 (pure water) | pH 5.4 (phosphate buffer) | 2-Propanol–pH 5.4 water, 1:1, (phosphate buffer) | ||
|---|---|---|---|---|---|
| Argon | Argon | O2 | Argon | ||
| I | ϕ ClPa | 0.038 ± 0.005 | 0.062 ± 0.007 | 0.004 ± 0.001 | 0.16 ± 0.015 |
| ϕ III | 0.027 ± 0.002 | 0.045 ± 0.004 | 0.002 ± 0.001 | 0.006 ± 0.003 | |
| ϕ Cl − | 0.038 ± 0.004 | — | — | — | |
| II | ϕ BrPa | — | 0.09 ± 0.01 | 0.08 ± 0.01 | — |
| ϕ III | — | 0.07 ± 0.01 | 0.07 ± 0.01 | — | |
3.3 Steady-state irradiation of II
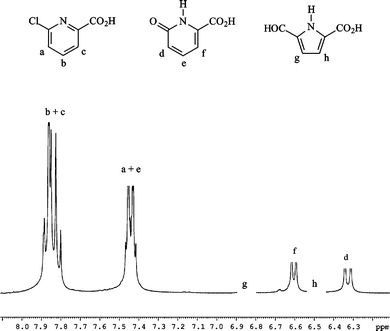
Irradiation of deoxygenated II (10−4 mol dm−3) gave the spectral changes shown in Fig. 3. The increase in absorbance within the wavelength range 290–350 nm was attributed to the formation of III. Another absorption band appeared at λ > 350 nm that was not detected in the case of aqueous I (see insert of Fig. 3). By HPLC analyses we could confirm the formation of III. We also detected compounds absorbing above 350 nm; these were found to have a sole molecular weight equal to 172 and are likely to be isomeric hydroxybipyridines (V). NMR spectra confirmed these attributions: the assignment of the signals of the phase sensitive COSY-45 is shown in Fig. 4. For the series of resonances belonging to the minor products, the shape of the broad signals fits well with the presence (<6%) of several hydroxybipyridines.17 The presence of substituted bipyridines in the reaction mixture is a clear indication that the scission of the C–Br bond was partly homolytic. The quantum yield of II photolysis (ϕBrPa) was 30% higher than that of I at pH 5.4 (see Table 1). III accounted for ≈ 80% of converted II. Oxygen did not affect photolysis of II, but, significantly inhibited the formation of V, as expected in the case of a formation via free radical chemistry.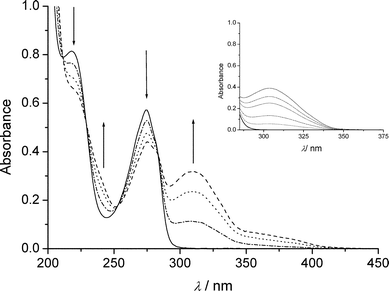 | ||
Fig. 3 Spectral changes of deoxygenated water solution of anionic II
(10−4 mol dm−3) upon irradiation at 280 nm: (—) initial; (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) )
t
= 17 min; (⋯)
t
= 40 min; ( )
t
= 17 min; (⋯)
t
= 40 min; (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) )
t
= 60 min. Insert: spectral changes for I in the same conditions, from bottom to top: initial; t
= 20 min; t
= 60 min; t
= 100 min; t
= 220 min; t
= 340 min. )
t
= 60 min. Insert: spectral changes for I in the same conditions, from bottom to top: initial; t
= 20 min; t
= 60 min; t
= 100 min; t
= 220 min; t
= 340 min. | ||
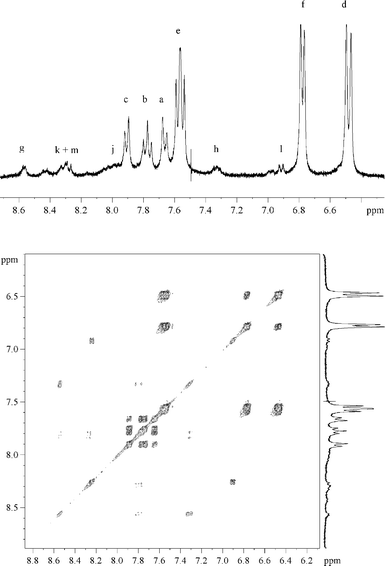 | ||
| Fig. 4 Proton COSY-45 of an irradiated solution of II. Conversion extent of 75%. A few drops of D2O were added to the initial solution in DMSO-d6. | ||
3.4 Laser flash photolysis
Fig. 5 shows transient absorption spectra observed from irradiated aqueous deoxygenated anionic I (pH 5.4). At pulse end, we obtained a broad band with a maximum at 305 nm and a shoulder around 375 nm. This absorption decayed by a first order kinetics (k = (2.8 ± 0.2) × 105 s−1) independently of I concentration. A stable absorption remained after the complete disappearance of the short-lived transient. It showed a maxima at 298 nm and a shoulder at 310 nm. It corresponded well to the overlapping of absorption spectra of III and IV. The short-lived transient was formed in a monophotonic process as indicated by the linear dependence of the absorbance change at 300 nm on the laser pulse energy. Using potassium peroxodisulfate as a chemical actinometer, we evaluated the product εϕ of the extinction coefficient and quantum yield from the slope of the linear regression to be 2200 ± 200 mol−1 dm3 cm−1 at 300 nm. In the oxygen-saturated medium the short-lived species readily disappeared with k = (2.1 ± 0.4) × 106 s−1 (insert A of Fig. 5). The rate constant of quenching by oxygen was estimated to be 1.4 × 109 dm3 mol−1 cm−1. Methyl acrylate (2 × 10−3 M) also reacted with the short-lived transient in deoxygenated medium; the rate constant was equal to 2.1 × 109 dm3 mol−1 cm−1 (insert B of Fig. 5). We thus assign the short-lived species to the triplet excited state. The absorption spectrum of I triplet resembled that of picolinic acid (Pa) triplet (λmax = 290 and 360 nm).17 In solutions containing oxygen or methyl acrylate, the secondary absorptions were not observed. The fact that oxygen scavenged the triplet excited state, inhibited I photolysis and suppressed the formations of III and IV demonstrates that the photochemistry of I arises from the triplet.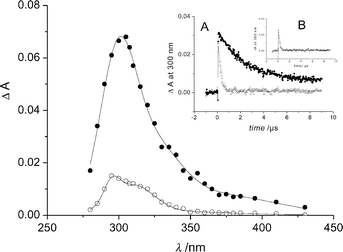 | ||
| Fig. 5 Transient spectra from deoxygenated anionic I measured at the end of the pulse (●) and 20 µs after the end of the pulse (○). Insert A: absorbance decay at 300 nm in deoxygenated medium (●) and in oxygen-saturated medium (△). Insert B: absorbance decay at 300 nm in deoxygenated medium containing methyl acrylate (2 × 10−3 mol dm−3). | ||
The formation of pyridine, 2-carboxypyridine and bipyridine derivatives from I in 2-propanol–water mixture strongly suggests a homolytic cleavage of the C–Cl bond. We tried to confirm the formation of chlorine atoms by irradiating I in the presence of chloride ions (0.01 mol dm−3) in the hope to produce Cl2−˙ radical ions (λmax
= 335 nm and ε
= 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dm3 mol−1 cm−1).22 Photolysis of I in a deoxygenated 2-propanol–water mixture in the absence of Cl− yielded the triplet again, as well as non-identified long-lived secondary absorptions. In the presence of Cl−
(0.01 mol dm−3) we observed an additional absorption within the wavelength range 310–380 nm bringing evidence for Cl2−˙ radical ions formation. We measured the absorbance change 6 µs after the pulse end at 335 nm, i.e., in conditions where the triplet had disappeared by more than 80%. As shown in Fig. 6, the increase in absorbance at 335 nm was linear with the laser energy pulse both in the absence and in the presence of NaCl (0.01 mol dm−3). Based on actinometry and after subtracting the slope obtained in the absence of NaCl from that obtained in its presence, we deduced that the product εϕ was 570 ± 150 dm3 mol−1 cm−1. The quantum yield of Cl2−˙ radical ion formation was thus equal to 0.057 ± 0.02.
000 dm3 mol−1 cm−1).22 Photolysis of I in a deoxygenated 2-propanol–water mixture in the absence of Cl− yielded the triplet again, as well as non-identified long-lived secondary absorptions. In the presence of Cl−
(0.01 mol dm−3) we observed an additional absorption within the wavelength range 310–380 nm bringing evidence for Cl2−˙ radical ions formation. We measured the absorbance change 6 µs after the pulse end at 335 nm, i.e., in conditions where the triplet had disappeared by more than 80%. As shown in Fig. 6, the increase in absorbance at 335 nm was linear with the laser energy pulse both in the absence and in the presence of NaCl (0.01 mol dm−3). Based on actinometry and after subtracting the slope obtained in the absence of NaCl from that obtained in its presence, we deduced that the product εϕ was 570 ± 150 dm3 mol−1 cm−1. The quantum yield of Cl2−˙ radical ion formation was thus equal to 0.057 ± 0.02.
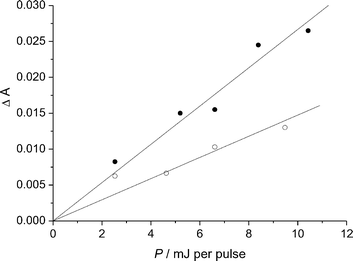 | ||
| Fig. 6 Absorbance change measured at 335 nm 6 µs after the end of the pulse as a function of the laser energy pulse (P) in a 2-propanol–water pH 5.4 (1 : 1) solution of I in the absence (○) and in the presence (●) of NaCl (0.01 mol dm−3). | ||
The photolysis of deoxygenated anionic II at 266 nm yielded at pulse end a broad and stable absorption with a maximum at 306 nm that was assigned to III. In addition, an absorption band was observed at λ > 350 nm. It was completed 5 µs after the pulse end. This latter absorption showed a maximum at 385 nm. Neither III, nor the 385 nm absorption was affected by the addition of oxygen in the medium. We acidified the solution of II to pH 1 in order to get the zwitterionic form. In these pH conditions, we could successfully detect a very short-lived species at pulse end along to III (Fig. 7). The short-lived species showed a spectrum reminiscent to that of I triplet with two maxima at 320 and 450 nm, but a much shorter lifetime (k > 3 × 107 s−1). It was attributed to the triplet of II.
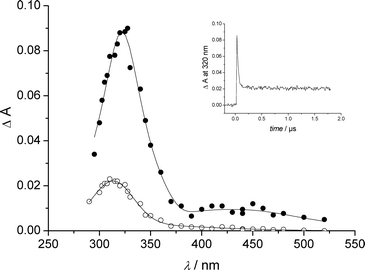 | ||
| Fig. 7 Transient spectra from deoxygenated water solution of II at pH 1 measured at the end of the pulse (●) and 2 µs after the end of pulse (○). Insert: absorbance decay at 320 nm. | ||
In the attempt to demonstrate that the C–Br bond was partly cleaved by a homolytic process, we photolyzed II in the presence of bromide ions (0.01 mol dm−3). This experiment allowed the detection of the characteristic broad absorption of Br2-radical ions (λmax
= 360 nm and ε
= 10000 dm3 mol−1 cm−1)22 in addition to that of the 385 nm transient. The absorbance measured 0.1 µs after the pulse end at 365 nm increased linearly with the laser pulse energy. Using chemical actinometry, we evaluated the product εϕ as being 260 ± 50 mol−1 dm3 cm−1 at 365 nm both in deoxygenated and oxygenated solutions. These values were corrected for the small contribution of the 385 nm transient produced in the absence of Br−
(less than 10%). Assuming a complete scavenging of Br˙ radicals by Br− we could deduce that the quantum yield of Br˙ formation was equal to 0.023 ± 0.005.
3.5 Mechanism of reaction
Results show that photodehalogenation of I is exclusively heterolytic in water and mainly homolytic in 2-propanol–water (1 : 1) mixture. Scheme 2 presents a mechanism that rationalizes these results. The triplet that is efficiently populated (εϕ = 2200 dm3 mol−1 cm−1) undergoes initial homolytic cleavage (process 1). A triplet radical pair consisting of 2-carboxypyridyl and Cl˙ radicals is formed. Due to the large atomic number of chlorine atom, the triplet radical pair should equilibrate with singlet radical pair (processes 2 and 3). From the singlet, geminate recombination (process d″) and electron transfer to yield an ion pair (process 4) can take place. The strong electron affinity of Cl˙ atoms (−3.61 eV atom−1)23 and the large dielectric constant of water are expected to favour electron transfer and make inefficient the free radical formation through cage escape either from the triplet or singlet radical pair. In solutions containing large proportions of 2-propanol, the electron transfer is likely to be more difficult due to the decrease of the dielectric constant of the medium. Consequently, the radical cage escape is favoured. Once formed, 2-carboxypyridyl radicals add to the starting compound yielding substituted bipyridines. They can also abstract a hydrogen atom from 2-propanol and produce 2-carboxypyridine or pyridine after decarboxylation. Subsequent to the electron transfer in pure water, ions will escape from the cage to yield halide ion and 2-carboxypyridinium. The latter is transformed into III after addition of water.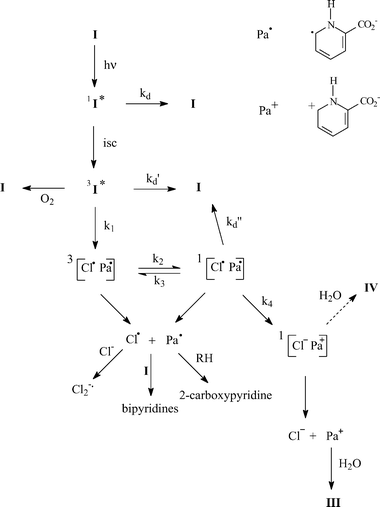 | ||
| Scheme 2 | ||
The formation of IV requires addition of H2O, elimination of HCl, and finally contraction of the ring. It only occurs from I. Thus, this product arises from an intermediate still bearing the halogen atom: the triplet or the ion pair. The complete inhibition of IV formation in 2-propanol–water mixture claims in favour of a formation by addition of water to the ion pair, before charge separation. Indeed, the generation of the ion pair through electron transfer becomes very minor upon addition of 2-propanol. Moreover, addition of water to the triplet should not be dramatically affected by 2-propanol when the latter is added at 50%. The pyrrole formation requires a ring contraction subsequently to HCl elimination and water addition. The detailed mechanism of this ring contraction is not clear.
To rationalize the data gained on II, we can suggest a mechanism similar to that shown in Scheme 2. The triplet excited state could not be either detected nor trapped at pH 5.4. Its involvement in the photoreaction cannot be therefore demonstrated, however, it is very likely. Indeed, due to internal heavy atom effect, brominated compounds are expected to populate triplet excited state as or more efficiently than chlorinated counterparts. We found that I populated the triplet excited state efficiently, thus, II triplet should be produced with a high yield too. Its non-observation may be due to a very short lifetime explained by a fast T → S intersystem crossing due to heavy atom effect again. Since the triplet is short-lived, the homolytic cleavage of the C–Br bond has to be a very fast reaction. If not, it cannot compete with deactivation. It should be much faster than homolytic cleavage of the C–Cl bond that arises from a long-lived triplet. The bond dissociation C–X is closely related to the C–X bond enthalpy. The latter is 60 kJ mol−1 lower for C6H5–Br than for C6H5–Cl.24 If we assume that similar differences exist for XPa, then the rate of the C–X homolytic cleavage is faster for II than for I. The detection of Br2−˙ upon irradiation of II in the presence of bromide ions provided evidence of the formation of bromine atoms. Bromine atoms may arise from homolytic cleavage of the C–Br bond but also from oxidation of Br− by II triplet. However, the latter reaction would produce the 6-bromo-2-carboxypyridinyl radical along with bromine atoms. We are sure that 2-carboxypyridinyl type radicals that are long-lived and easy to detect were not produced in our system.17 Thus bromine atoms arise mainly from IIvia homolytic cleavage of the C–Br bond. In the mechanism shown in Scheme 2, the radical formation competes with the electron transfer. The electron affinity of Br˙ atoms (−3.36 eV atom−1 )23 is lower than that of Cl˙ atoms. The rate of electron transfer for II is thus expected to be smaller than that of I in good accordance with a higher efficiency of radical formation. The last particularity of II is that bipyridines are decarboxylated. This implies that decarboxylation of radicals is very efficient: the high efficiency of 2-carboxypyridyl radical decarboxylation was demonstrated in a previous work.17
In conclusion, the photochemistry of two halogenocarboxypyridines, I and II, in water has been elucidated. Heterolytic cleavage of the C–X bond is observed for I in water, while both homolytic and heterolytic cleavages occur for II in water and for I in a 2-propanol–water (1 : 1) mixture. The triplet excited state of I was observed (λmax = 305 nm) and its involvement in the reaction demonstrated on the basis of scavenging experiments. Initial homolytic cleavage of the C–X bond from the triplet to form a radical pair may be followed by an electron transfer from carboxypyridyl radical to the halogen atom that competes with radical cage escape. Thus, the heterolytic vs. homolytic pathway is governed by the reactivity of the radical pair, depending on the electron affinity of the halogen and on the polarity of the medium.
References
- L. O. Ruzo, M. J. Zabik and R. D. Schuetz, Photochemistry of bioactive compounds. Photochemical processes of polychlorinated biphenyls, J. Am. Chem. Soc., 1974, 96, 3809–3813 CrossRef CAS
.
- N. J. Bunce, P. Pilon, L. O. Ruzo and D. J. Sturch, Electron transfer on photolysis of 1-chloronaphthalene in alkane solvents, J. Org. Chem., 1976, 41, 3023–3025 CrossRef CAS
- P. J. Wagner and B. J. Scheve, Steric effects in the singlet–triplet transitions of methyl- and chlorobiphenyls, J. Am. Chem. Soc., 1977, 99, 2888–2892 CrossRef CAS
- D. R. Arnold and P. C. Wong, The photochemistry of chloroaromatic compounds. Is “π-chlorobenzene” an intermediate?, J. Am. Chem. Soc., 1977, 99, 3361–3366 CrossRef CAS
- J. P. Soumillion and B. De Wolf, A link between photoreduction and photosubstitution of chloroaromatic compounds, J. Chem. Soc., Chem. Commun., 1981, 436–437 RSC
- Y. Yamamoto, T. Aoyama and K. Hayashi, Pulse radiolysis study of salt effects on reactions of aromatic radical cations with Cl−, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2209–2214 RSC
- N. J. Bunce, J. P. Bergsma, M. D. Bergsma, W. De Graaf, Y. Kumar and L. Ravanal, Structure and mechanism in the photoreduction of aryl chlorides in alkane solvents, J. Org. Chem., 1980, 45, 3708–3713 CrossRef CAS
- T. Moore and R. M. Pagni, Unusual photochemistry of 4-chlorobiphenyl in water, J. Org. Chem., 1987, 52, 770–773 CrossRef CAS
- J. Orvis, J. Weiss and R. M. Pagni, Further studies on the photoisomerization and hydrolysis of chlorobiphenyls in water. Common ion effect in the photohydrolysis of 4-chlorobiphenyl, J. Org. Chem., 1991, 56, 1851–1857 CrossRef CAS
- G. Zhang and P. Wan, Photogeneration of HF from fluoromethoxybenzenes in aqueous solution, J. Chem. Soc., Chem. Commun., 1994, 19–20 RSC
- P. J. Kropp, G. S. Poindexter, N. J. Pienta and D. C. Hamilton, Photochemistry of alkyl halides. 4. 1-norbornyl, 1-norbornylmethyl, 1- and 2-adamantyl, and 1-octyl bromides and iodides, J. Am. Chem. Soc., 1976, 98, 8135–8144 CrossRef CAS
- H. Ishihara and S. Yi Wang, Photochemistry of 5-bromouracil in aqueous solution, Biochemistry, 1966, 5, 2307–2313 CrossRef CAS
- H. Ishihara and S. Yi Wang, Photochemistry of 5-bromouracils: isolation of 5,5′-diuracils, Nature, 1966, 210, 1222–1225 CAS
- L. R. Hamilton and P. J. Kropp, Phototransformation of 2,4-dimethoxy-6-chloropyrimidine, Tetrahedron Lett., 1971, 12, 1625–1628 CrossRef
- B. J. Swanson, J. C. Kutzer and T. H. Koch, Photoreduction of 5-bromouracil. Ionic and free-radical pathways, J. Am. Chem. Soc., 1981, 103, 1274–1276 CrossRef CAS
- L. Lindqvist, B. Czochralska, M. P. Fontaine-Aupart, W. Kawczynski, F. Tfibel and T. Douki, Photochemistry of 2-chloropyrimidine, Photochem. Photobiol. Sci., 2002, 1, 600–606 RSC
- F. Rollet, C. Richard, J. F. Pilichowski and B. Aboab, Photochemistry of the three carboxypyridines in water: a pH dependent reaction, Org. Biomol. Chem., 2004, 2, 2253–2261 RSC
- F. Bonnichon and C. Richard, Phototransformation of 3-hydroxybenzonitrile in water, J. Photochem. Photobiol., A, 1998, 119, 25–32 CrossRef CAS
- G. Grabner, personal communication.
- Calculated using Advanced Chemistry Development (ACD/Labo) software Solaris V4.67, 1994–2005, data base of Chemical Abstracts Service Search PubMed
- R. W. Green and H. K. Tong, The constitution of the pyridine monocarboxylic acids in their isoelectric forms, J. Am. Chem. Soc., 1956, 78, 4896–4900 CrossRef
- P. Neta, R. E. Huie and A. B. Ross, Rate constants for reactions of inorganic radicals in aqueous solution, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CAS
-
T. Miller, Handbook of Chemistry and Physics, ed D. R. Lide, CRC Press, Boca Raton, 85th edn, 2004–2005, section
10, p. 148 Search PubMed
-
J. Kerr, Handbook of Chemistry and Physics, ed D. R. Lide, CRC Press, Boca Raton, 85th edn, 2004–2005, section 9, p. 75 Search PubMed
| This journal is © The Royal Society of Chemistry and Owner Societies 2006 |