Photorelease of carboxylic and amino acids from N-methyl-4-picolinium esters by mediated electron transfer
Received 8th August 2005, Accepted 14th October 2005
First published on 16th November 2005
Abstract
One electron reduction of N-alkyl-4-picolinium (NAP) esters initiates C–O bond scission releasing a carboxylate anion. Previous experiments have demonstrated that this process can be initiated by photoinduced electron transfer from an electron-donating sensitizer. In the present study it is demonstrated that a comparable photorelease process can be initiated by photolysis of an electron acceptor (mediator), which in turn abstracts an electron from a ground state electron donor. The resulting mediator anion radicals donate an electron to the NAP ester, triggering release of the carboxylate anion. It is demonstrated that when benzophenone is used as a mediator, higher quantum yields for ester decomposition can be achieved compared with sensitizers that do direct photoinduced electron transfer.
1. Introduction
There has been much recent interest in the development of improved photoremovable protecting groups (PRPG) due to their expanding applications in endeavors such as photolithography,1 molecular switches,2 spatio–temporal modulation of cellular processes,3,4 and organic synthesis.5 Most PRPG are designed to absorb light directly, i.e. the chromophore is intrinsic to the protecting group. This means that any synthetic modifications aimed at optimizing the light absorbing properties of the PRPG invariably alter the rates and efficiencies of the desired release process. Examples of PRPG with intrinsic chromophores include the well known ortho-nitrobenzyl esters and ethers,6,7 as well as more recently characterized PRPG such as those based on ortho-alkylarylketones (e.g. 2,6 dimethylphenacyl, or DMP group),8,9 benzoin ester derivatives,10,11para-hydroxyphenacyl (p-HP groups) esters,12,13 coumarin,14,15 and 7-nitroindole photochemistry (NI group).16,17An alternative strategy is to activate photorelease using an extrinsic chromophore, or photosensitizer. The latter is designed to activate the protecting group by means of an excited state energy or electron transfer step. By varying the photosensitizer, it is possible to optimize the wavelength response of the system without affecting the rates or mechanism of the release step. Examples of PRPG with extrinsic chromophores include Kutateladze et al.'s dithiane based PRPG,18 Yonemitsu et al.'s DNMBS group19 and Corrie et al.'s 7-NI groups linked to triplet sensitizers.20,21 Our work has focused on two such PRPG: the phenacyl group (Pac)22 and the N-alkyl-4-picolinium (NAP) group.23 Both of these can be released through photoinduced electron transfer from an extrinsic sensitizer. The NAP group, in particular, can be activated with wavelengths as high as 525 nm, using visible light-absorbing laser dyes (Scheme 1).24
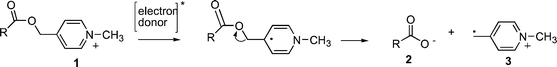 |
| | Scheme 1 C–O Bond fragmentation in NAP esters triggered by one electron reduction. | |
Most PRPG applications require high quantum yields for substrate release (Φrel) In general, the release quantum yields for PET activated PRPG is limited by non productive back electron transfer reactions, (kBET) which compete with bond breaking (kfrag) or separation of the resulting ion radical pair through diffusion (kesc) as shown in Scheme 2. In the case of the NAP esters, the highest Φrel are as high as 20%, in cases where a separate sensitizer is used.23 When the sensitizer is covalently attached to the NAP group, the Φrel is negligible, presumably because krel
≪
kBET.25 This caused us to examine alternative methods for activating the NAP group.
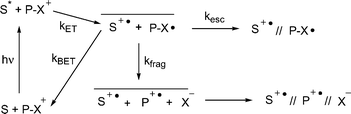 |
| | Scheme 2 Direct photosensitized fragmentation of NAP esters (1) using excited state electron donors. | |
Photoinduced electron transfer using a mediator (or co-sensitizer) has been shown to substantially increase the quantum yields of such reactions when compared with direct PET sensitization.26,27 In a mediated PET scheme, an excited state mediator abstracts an electron from a ground state donor (Scheme 3). The resulting reduced mediator then donates an electron to the PRPG. Because the second electron transfer occurs in the ground state and is sufficiently exergonic to be practically irreversible, the competition between kBET and krel is absent. Of course there is such a competition in the primary electron transfer step, but this can be designed such that (a) the kBET is in the Marcus inverted region, and thus slow or (b) occurs through a triplet excited state, producing a triplet geminate radical pair, where kBET is a spin-forbidden process.
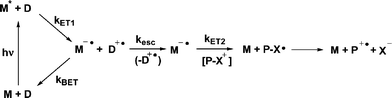 |
| | Scheme 3 Activation of NAP esters by mediated photoinduced electron transfer. | |
In a preliminary report, we described the photochemistry of a covalently linked mediator–PRPG molecule (4, Scheme 4).25 Upon photolysis in the presence of an electron donor (N,N-dimethylaniline, DMA), the benzophenone chromophore on this compound abstracts an electron, and then (apparently) transfers the electron to the NAP group, triggering a scission of the C–O bond, and releasing the carboxylate group with Φrel = 0.72. Comparable directly sensitized linked systems (using a 9-carbazolyl group as the electron donor (5)) show no measurable reactivity (Φrel < 0.001). This article elaborates on this preliminary report. Specifically results with several unlinked mediator–NAP combinations are examined.
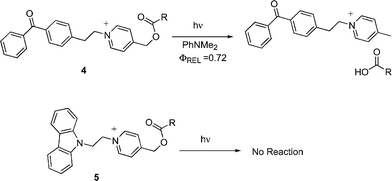 |
| | Scheme 4 Mediator and sensitizer linked NAP esters. | |
2. Results and discussion
(a) Design considerations
The selection of mediators for this study was guided by several considerations. First, the mediators should absorb light at >340 nm to avoid competing absorption by the protected molecules. Second, in order to carry out the desired electron transfer processes, kET1 and kET2, the mediators need to have reduction potentials sufficiently negative to reduce the NAP group, but not so negative that they cannot be reduced in the photochemical step. The mediators employed in these studies include benzophenone (BP), xanthone (XA) and 9, 10-diphenylanthracene (DPA) (Chart 1). The former two sensitizers are characterized by rapid rates of intersystem crossing (ISC). Thus, it is expected that the PET reactions of these species will originate from their triplet states. On the other hand DPA has a low efficiency of intersystem crossing and is thus expected to carry out PET from its singlet state. However, the high exergonicity predicted for BET (−56 kcal mol−1) suggested that BET for this mediator should occur in the Marcus inverted region and be slow relative to solvent cage escape. |
| | Chart 1 Mediators used in this study. | |
Several key photophysical and electrochemical parameters for the mediators are supplied in Table 1. These include the highest wavelength absorption maximum (λmax), the reduction potential (Ered), and the excited state energy (E0–0) of the mediators. From these parameters, we calculated the driving force for the initial PET reaction from the ground state donor N,N-dimethylaniline (DMA) using its published oxidation potential (EDMA) of 0.53 V vs. SCE, and eqn (1). In that expression, the factor of 0.06 eV is a solvent term. The driving force for the back electron transfer step (ΔGBET) is predicted from eqn (2) and that for the secondary electron transfer (ΔGET2) is predicted from eqn (3), where EPRPG is the reduction potential of the NAP group (−1.1 V vs. SCE).
| | | ΔGPET = 23.06 (EM
−
EDMA
−
E0–0 + 0.06) | (1) |
| | | ΔGBET = 23.06 (EDMA
−
EM) | (2) |
| | | ΔGET2 = 23.06 (EM
−
EPRPG) | (3) |
Table 1 Photophysical and redox properties of the mediators28,29
| Mediator | λmax/nm | Ered/V | E0-0 (s)/eVa | E0-0 (t)/eVb | ΔGPET/kcal mol−1 | ΔGBET/kcal mol−1 | ΔGET2/kcal mol−1 |
|---|
| Singlet state. Triplet state. |
|---|
| XA | 340 | −1.77 | 3.36 | 3.21 | −13.1 | −22.1 | −15.4 |
| BP | 384 | −1.68 | 3.23 | 3.01 | −10.6 | −20.1 | −13.3 |
| DPA | 392 | −1.94 | 3.16 | — | −9.45 | −63.4 | −19.4 |
(b) Preparation and mediated photolysis of NAP esters
Five carboxylic acids were protected as NAP esters. These esters were synthesized as described previously, and their structures are shown in Chart 2.23,24 They include esters of three simple carboxylic acids: phenylacetic acid 1a, diphenylacetic acid 1b, and 4-tolylacetic acid 1c. Also examined were NAP esters of N-carbonylbenzyloxy (cbz) protected glycine 1d and serine 1e. |
| | Chart 2 N-Methyl-4-picolinium esters used in this study. | |
The anticipated C–O bond scission was effected by photolyzing deoxygenated 2 mL solutions consisting of the NAP ester 1 (4 mM), mediator (2 mM), and excess of the electron donor, DMA using 200 W Hg arc lamp with a 320 nm cut-off filter. Following photolysis for the prescribed times, the solvent was removed under vacuum, and the sample was dissolved in deuterated solvent containing an internal standard. The acid and the ester were then quantified by comparing their peak integrations of the α-C–H to that of the internal standard. In all cases 1H NMR spectra of the product mixtures showed clean conversion of the NAP esters into the corresponding carboxylic acids along with N-methylpicolinium ion. As indicated in Table 2, the carboxylic acid product accounts for all, or nearly all, of the consumed NAP ester. In each case a nearly equimolar amount of N-methyl-4-picolinium ion was also observed in the 1H NMR spectra of the photolysis mixtures. For several examples, indicated in Table 2, the assignment of the product was confirmed by isolating the carboxylic acids. In these cases the yields were determined gravimetrically and the identities of the carboxylic acids were confirmed by matching their 1H NMR spectra to those of authentic standards.
Table 2 Photorelease of NAP esters in the benzophenone–DMA system
| Mediator | Ester | Conditionsa | Acid formed (%)b,c | Ester consumed (%)b,c |
|---|
| Photolysis solution was 2.5–3.0 mL of 2 mM mediator, 4 mM 1 and 10–15 mM DMA. Yields were determined by 1H NMR integration of the acid peak relative to an internal standard. Estimated error ±5%. Quantities are 3-fold higher than indicated in (a) and yields were determined by isolating and weighing the product. |
|---|
| Benzophenone | 1a | 2 h, MeOH | 94 | 94 |
| Benzophenone | 1a | 10 h, MeOHd | 84 | 100 |
| Benzophenone | 1b | 2 h, MeOH | 84 | 88 |
| Benzophenone | 1c | 2 h, MeOH | 78 | 80 |
| Benzophenone | 1d | 2 h, MeOH | 97 | 97 |
| Benzophenone | 1e | 2 h, MeOH | 100 | 100 |
| Xanthone | 1a | 3 h, MeOH | 62 | 62 |
| Xanthone | 1a | 15 h MeOHd | 74 | 100 |
| Xanthone | 1b | 3 h, MeOH | 86 | 84 |
| Xanthone | 1c | 3 h, MeOH | 73 | 80 |
| Xanthone | 1d | 3 h, MeOH | 88 | 88 |
| Xanthone | 1e | 3 h, MeOH | 100 | 100 |
| DPA | 1a | 3 h, MeCN–MeOH | 98 | 100 |
| DPA | 1a | 15 h, MeCN–MeOH | 81 | 100 |
| DPA | 1b | 3 h, MeCN–MeOH | 100 | 100 |
| DPA | 1c | 3 h, MeCN–MeOH | 81 | 85 |
| DPA | 1d | 3 h, MeCN–MeOH | 100 | 100 |
| DPA | 1e | 3 h, MeCN–MeOH | 94 | 100 |
Control experiments were carried out wherein DMA, the electron donor, was omitted. With DPA and XA, <10% conversion of the starting material was observed. With BP slightly larger amounts (<20%) were observed in the control experiments. The latter are attributed to reactions of the photogenerated benzophenone ketyl radical (Ph2COH˙), which forms upon H atom abstraction from the solvent.
(c) Quantum yields
Experimental values for Φrel were determined by irradiating a solution of mediator, DMA and picolinium ester with a 1000 W xenon lamp, fitted with a monochromator set to the absorption maxima of the mediator and a bandwidth of 10 nm. The quantum yields for the mediated systems were found to be significantly greater than the direct sensitized cases. The quantum yield for photofragmentation of picolinium esters by direct sensitization with 9-methylcarbazole was determined earlier to be 0.23 under similar conditions.23 Also, the quantum yield of the mediated DPA–DMA system was found to be only 0.17, which is much lower than the quantum yields observed for the two triplet mediated systems. Evidently, the triplet-mediated electron transfer proceeds more efficiently than direct sensitization or singlet-mediated electron transfer (Table 3).
Table 3 Quantum yields for mediated photofragmentation of NAP ester 1a
| Donor | Mediator | Ester | Conditions | Φrel |
|---|
| DMA | Benzophenone | 1a | MeOH | 0.51 |
| DMA | Xanthone | 1a | MeOH | 0.39 |
| DMA | DPA | 1a | MeCN–MeOH | 0.17 |
(d) Laser flash photolysis experiments
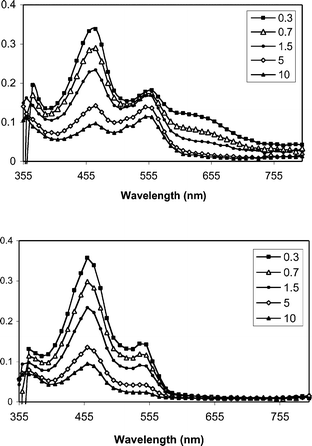
The proposed mediated PET mechanism is supported by laser flash photolysis experiments. Fig. 1–3 shows transient spectra from pulsed laser (355 nm, 10 ns, 20 mJ) photolysis of solutions containing one of the three mediators and DMA (upper panels) in methanol, along with similar solutions that include NAP ester 1b (lower panels). In the case of XA (Fig. 1), photolysis in the presence of DMA provides the expected radical ion intermediates: DMA+˙ (460 nm) and XA−˙ (560 nm).30,31 When 1b is included, the XA−˙ signal is abolished, but the DMA+˙ signal remains.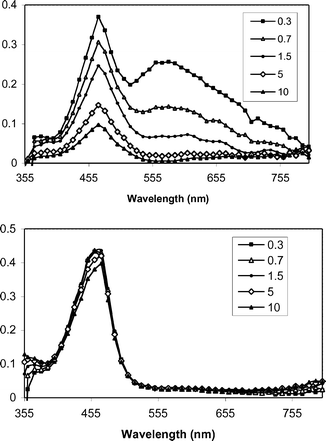 |
| | Fig. 1 Transient spectra from pulsed laser photolysis (355 nm, 50–100 mJ, 4–6 ns) of XA + DMA (top) and XA + DMA+ 1b in N2-purged CH3OH. The inset indicates the time (µs) following the excitation pulse. | |
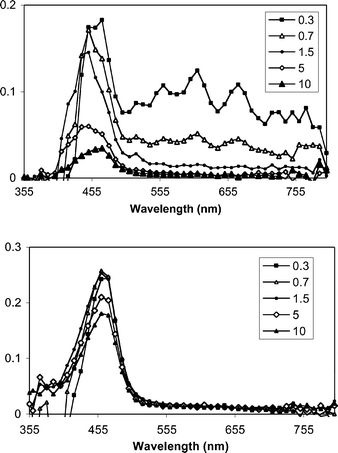 |
| | Fig. 2 Transient spectra from pulsed laser photolysis (355 nm, 50–100 mJ, 4–6 ns) of DPA + DMA (top) and DPA + DMA+ 1b in N2-purged CH3CN–CH3OH mixtures. The inset indicates the time (µs) following the excitation pulse. | |
 |
| | Fig. 3 Transient absorption spectra from pulsed photolysis (355 nm, 50–100 mJ, 4–6 ns) of BP + DMA (top) and BP + DMA + NAP ester 1b (bottom) in N2-purged CH3OH. The inset indicates the time (µs) following the excitation pulse. | |
Electron transfer from XA−˙ to 1b occurs near the diffusion limit in CH3OH. The decay rate constant for XA−˙ was characterized at various concentrations of 1b and a pseudo-first order analysis of these data (Fig. 4) provides a value of 9 × 109 M−1 s−1 for kET2.
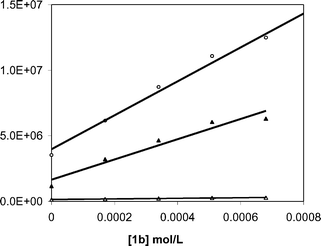 |
| | Fig. 4 Dependence of the decay rate constants for BP−˙ (open circles), XA−˙ (filled triangles) and BPH˙ (open triangles) on the concentration of NAP ester 1b. | |
With DPA, similar results are obtained. LFP experiments with DPA and DMA provide transient signals for DMA+˙ and DPA−˙ (560, 610 and 670 nm). The latter signals are rapidly eliminated upon addition of 1b, whereas DMA+˙ is not quenched. In fact, its lifetime increases when 1b is added. The latter effect is attributed to the consumption of DPA−˙, which in the absence of 1b, serves as the main pathway for the loss of DMA+˙.
In the case of BP, the transient behavior is a bit more complicated. Previous studies have shown that the initial electron transfer from DMA to triplet benzophenone is followed by two competing processes: solvent cage escape, and proton transfer from DMA+˙ to BP−˙. In methanol, these processes occur with comparable rates. Thus, process kET1 generates a combination of BP−˙ and its conjugate acid, BPH˙ (Scheme 5). Our experiments show results (Fig. 3) that are consistent with this picture.
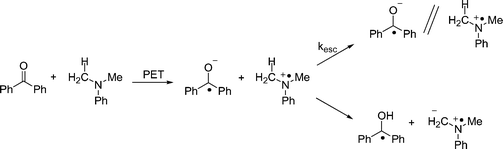 |
| | Scheme 5 | |
As shown in Fig. 3, addition of 1b eliminates the BP−˙ signal and diminishes the lifetime of BPH˙. As with xanthone, quenching experiments were carried out for both of the reduced benzophenone species (Fig. 4) and it was found that BP−˙ reacts at or near the diffusion limit with 1b, kET2 = 1 × 1010 M−1 s−1, but that BPH˙ reacts more slowly with 1b, with kET2 = 3 × 108 M−1 s−1. We assume the latter process consists of a coupled electron transfer and deprotonation reaction. However its mechanism has yet to be examined in detail. Nonetheless, in view of the high Φrel for the benzophenone mediated photorelease, it seems likely that both pathways contribute to the C–O bond scission.
3. Conclusions
Photofragmentation of NAP esters via mediated photoinduced electron transfer reactions offers the advantages of higher quantum yields compared to situations where the electron transfer step occurs directly from excited photosensitizers. The unlinked mediator–NAP ester photochemistry examined in this study does not show Φrel values that are improved over comparable covalently linked systems, 4. However, the current systems are attractive because they do not require the additional synthetic steps of linking the mediator to the NAP group. On the other hand, as this approach requires interaction of three separate molecules, it is likely to be practical only in situations where the donor, ester, and mediator concentrations can be kept in the millimolar range.4 Experimental
(a) General procedures
All 1H and 13C NMR were obtained using a Bruker 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) in hertz (Hz). Deuterated CD3CN or CD3OD were used for all NMR experiments. Acetonitrile was distilled from calcium hydride under nitrogen atmosphere. Ultraviolet-visible spectra were recorded on a Perkin-Elmer Lambda 2S spectrophotometer.(b) Deprotection photolysis
A solution of 4–10 mg of the picolinium ester, 2–4 mg of mediator and an excess of donor DMA was prepared in about 5 mL of CH3OH (2 mM of mediator, 4 mM of ester and 10–15 mM of DMA). Half of the solution was used as dark control and the remaining half was purged with nitrogen, and irradiated using a 200 W Hg lamp with continuous stirring. A pyrex filter was used to cut-off light below 325 nm to ensure light absorption by the mediator alone. The solutions were then evaporated and redissolved in CD3OD with hexamethyldisiloxane as the internal standard and 1H NMR spectra were obtained. The percent yields were determined by 1H NMR integration of the carboxylic acid peaks relative to the internal standard. The estimated error is ±5%.(c) Preparative photolysis
The preparative photolysis mixture was scaled up by a factor of 3, from the deprotection photolysis, and the irradiation time was scaled up by a factor of 5 to ensure complete conversion of the ester. The solution was N2-purged and photolyzed using a 200 W Hg lamp (with a 325 nm cut-off filter) with continuous stirring. The photolyzed solution was evaporated, and the residue dissolved in CH2Cl2 (20 mL). The free acid was extracted into aqueous layer by addition of NaHCO3 (4 × 10 mL). The aqueous layer was acidified and extracted with CH2Cl2 (4 × 10 mL). The organic layer was dried over MgSO4, and concentrated in vacuo. The product was weighed and analyzed by 1H NMR and compared against the standard.(d) Laser flash photolysis
Laser flash photolysis experiments were performed using an Nd:YAG laser as the pump beam source. The laser used was a Continuum Surelite II-10 capable of 266 or 355 nm pulses between 4–6 ns duration. A LeCroy 350 MHz digital oscilloscope was used to observe the waveforms. The samples were prepared such that the absorbances were between 1.5 and 2.0 at the excitation wavelength, 355 nm. The samples were placed in 1 cm quartz cuvettes and N2 (or O2) purged for 15 min, and stirred continuously during the photolysis.(e) Quantum yield determination
The solutions were photolyzed by the output of a 1000 W Hg–Xe lamp after passing through a spectral energy monochromator set to the absorption maxima of the mediators with a 10 nm bandwidth. Solutions of ester, mediator and DMA (8 mM ester, solution OD at λmax about 0.1–0.3) were placed in a 1 cm quartz cuvette and purged with N2 for 10–15 min. The samples were irradiated for different times and the amount of ester converted was determined by 1H NMR integration. Light intensities were measured by a radiometer calibrated by ferrioxalate actinometry.Acknowledgements
Financial support was provided by the NSF. We thank Prof. N. Blough for assistance with quantum yield determinations.References
- M. C. Pirrung, L. Fallon and G. McGall, Proofing of photolithographic DNA synthesis with 3′,5′-dimethoxybenzoincarbonyl-protected deoxynucleoside phosphoramidites, J. Org. Chem., 1998, 63, 241–246 CrossRef CAS.
- M. C. Pirrung, W. H. Pieper, K. P. Kaliappan and M. R. Dhananjeyan, Combinatorial discovery of two-photon photoremovable protecting groups, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12548–12553 CrossRef CAS.
- G. Marriot and J. W. Walker, Caged peptides and proteins: New probes to study polypeptide function in complex biological systems, Trends Plant Sci., 1999, 4, 330–334 CrossRef.
- A. Heckel and G. Mayer, Light regulation of aptamer activity: An anti-thrombin aptamer with caged thymidine nucleobases, J. Am. Chem. Soc., 2005, 127, 822–823 CrossRef CAS.
- V. N. R. Pillai, Photoremovable protecting groups in organic synthesis, Synthesis, 1980, 1–26 CAS.
- Y. V. Il'ichev, M. A. Schworer and J. Wirz, Photochemical reaction mechanisms of 2-nitrobenzyl compounds: Methyl ethers and caged ATP, J. Am. Chem. Soc., 2004, 126, 4581–4595 CrossRef CAS.
- C. Grewer, J. Jager, B. K. Carpenter and G. P. Hess, A new photolabile precursor of glycine with improved properties: A tool for chemical kinetic investigations of the glycine receptor, Biochemistry, 2000, 39, 2063–2070 CrossRef CAS.
- J. Literak, J. Wirz and P. Klan, 2,5-Dimethylphenacyl carbonates: A photoremovable protecting group for alcohols and phenols, Photochem. Photobiol. Sci., 2005, 4, 43–46 RSC.
- P. Klán, A. P. Pelliccioli, T. Pospisil and J. Wirz, 2,5-Dimethylphenacyl esters: A photoremovable protecting group for phosphates and sulfonic acids, Photochem. Photobiol. Sci., 2002, 1, 920–923 RSC.
- R. S. Rock and S. I. Chan, Preparation of a water-soluble “cage” based on 3′,5′-dimethoxybenzoin, J. Am. Chem. Soc., 1998, 120, 10766–10767 CrossRef CAS.
- C. S. Rajesh, R. S. Givens and J. Wirz, Kinetics and mechanism of phosphate photorelease from benzoin diethyl phosphate: Evidence for adiabatic fission to an α-keto cation in the triplet state, J. Am. Chem. Soc., 2000, 122, 611–618 CrossRef CAS.
- R. S. Givens, A. Jung, C.-H. Park, J. Weber and W. Bartlett, New photoactivated protecting groups. 7. p-hydroxyphenacyl: A phototrigger for excitatory amino acids and peptides, J. Am. Chem. Soc., 1997, 119, 8369–8370 CrossRef CAS.
- R. S. Givens, J. F. W. Weber, P. G. Conrad, G. Orosz, S. L. Donahue and S. A. Thayer, New phototriggers 9: p-Hydroxyphenacyl as a C-terminal photoremovable protecting group for oligopeptides, J. Am. Chem. Soc., 2000, 122, 2687–2697 CrossRef CAS.
- T. Furuta, H. Torigai, M. Sugimoto and M. Iwamura, Photochemical properties of new photolabile cAMP derivatives in a physiological saline solution, J. Org. Chem., 1995, 60, 3953–3956 CrossRef CAS.
- T. Furuta, S. S.-H. Wang, J. L. Dantzker, T. Dore, E. J. Bybee, E. M. Callaway, W. Denk and R. Y. Tsien, Brominated 7-hydroxycoumarin-ylmethyls: Photolabile protecting groups with useful cross-sections for two-photon photolysis, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1193–2000 CrossRef CAS.
- G. Papageorgiou, D. C. Ogden, A. Barth and J. E. T. Corrie, Photorelease of carboxylic acids from 1-acyl-7-nitroindolines in aqueous solution: Rapid and efficient photorelease of L-glutamate, J. Am. Chem. Soc., 1999, 121, 6503–6504 CrossRef CAS.
- M. Canepari, L. Nelson, G. Papageorgiou, J. E. T. Corrie and D. C. Ogden, Photochemical and pharmacological evaluation of 7-nitroindolinyl- and 4-methoxy-7-nitroindolinyl-amino acids as novel, fast caged neurotransmitters, J. Neurosci. Methods, 2001, 112, 29–42 CrossRef CAS.
- Z. Li, Y. Wan and A. G. Kutateladze, Dithiane-based photolabile amphiphiles: Toward photolabile liposomes, Langmuir, 2003, 19, 6381–6391 CrossRef CAS.
- T. Hamada, A. Nishida and O. Yonemitsu, A new amino protecting group readily removable with near
ultraviolet light as an application of electron-transfer photochemistry, Tetrahedron Lett., 1989, 30, 4241–4244 CrossRef CAS.
- G. Papageorgiou, D. C. Ogden and J. E. T. Corrie, An antenna-sensitized nitroindoline precursor to enable photorelease of L-glutamate in high concentrations, J. Org. Chem., 2004, 69, 7228–7233 CrossRef CAS.
- G. Papageorgiou, M. Lukeman, P. Wan and J. E. T. Corrie, An antenna triplet sensitiser for 1-acyl-7-nitroindolines improves the efficiency of carboxylic acid photorelease, Photochem. Photobiol. Sci., 2004, 3, 366–373 RSC.
- A. Banerjee and D. E. Falvey, Protecting groups that can be released through photoinduced electron transfer: Mechanistic and product studies of photosensitized release of carboxylates from phenacyl esters, J. Org. Chem., 1997, 62, 6245–6251 CrossRef CAS.
- C. Sundararajan and D. E. Falvey, C–O bond fragmentation of 4-picolyl and N-methyl-4-picolinium esters triggered by photochemical electron transfer, J. Org. Chem., 2004, 69, 5547–5554 CrossRef CAS.
- C. Sundararajan and D. E. Falvey, Photorelease of carboxylic acids, amino acids, and phosphates from N-alkylpicolinium esters using photosensitization by high wavelength laser dyes, J. Am. Chem. Soc., 2005, 127, 8000–8001 CrossRef CAS.
- C. Sundararajan and D. E. Falvey, Photolytic release of carboxylic acids using linked donor–acceptor molecules: Direct versus mediated photoinduced electron transfer to N-alkyl-4-picolinium esters, Org. Lett., 2005, 7, 2631–2634 CrossRef CAS.
- T. L. Morkin, N. J. Turro, M. H. Kleinman, C. S. Brindle, W. H. Kramer and I. R. Gould, Selective solid state photooxidant, J. Am. Chem. Soc., 2003, 125, 14917–14924 CrossRef CAS.
- H. Rinderhagen and J. Mattay, Cyclopropyl silyl ether radical cations. Part 1: Synthetic applications in radical/radical cationic cascade reactions, Chem.–Eur. J., 2004, 10, 851–874 CrossRef CAS.
- S. L. Murov, I. Carmichael, and G. L. Hug, Handbook of photochemistry, Marcel Dekker Inc., New York, 2nd edn, 1993 Search PubMed.
- A. J. Bard, K. S. V. Santhanam, J. T. Maloy, J. Phelps and L. O. Wheeler, Steric effects and the electrochemistry of phenyl-substituted anthracenes and related compounds, Discuss. Faraday Soc., 1968, 45, 167–174 RSC.
- T. Shida, Electronic absorption spectra of radical ions, Elsevier, Amsterdam, 1988 Search PubMed.
- M. Goez and B. H. M. Hussien, Photoionization of xanthone via its triplet state or via its radical anion, Phys. Chem. Chem. Phys., 2004, 6(24), 5490–5497 RSC.
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2006 |
Click here to see how this site uses Cookies. View our privacy policy here.