Integrated thin-film polymer/fullerene photodetectors for on-chip microfluidic chemiluminescence detection
Xuhua
Wang
ac,
Oliver
Hofmann
bc,
Rupa
Das
a,
Edward M.
Barrett
a,
Andrew J.
deMello
bc,
John C.
deMello
*bc and
Donal D. C.
Bradley
*ac
aExperimental Solid State Physics Group, Blackett Laboratory, Imperial College London, United Kingdom SW7 2AZ. E-mail: d.bradley@imperial.ac.uk
bElectronic Materials Group, Department of Chemistry, Imperial College London, United Kingdom SW7 2AZ. E-mail: j.demello@imperial.ac.uk
cMolecular Vision Ltd, Imperial BioIncubator, Imperial College London, United Kingdom SW7 2AZ
First published on 17th October 2006
Abstract
We report the use of solution-processed thin-film organic photodiodes for microscale chemiluminescence. The active layer of the photodiodes comprised a 1 : 1 blend by weight of the conjugated polymer poly(3-hexylthiophene) [P3HT] and [6,6]-phenyl-C61-butyric acid-methylester [PCBM]—a soluble derivative of C60. The devices had an active area of 1 mm × 1 mm, and a broad-band response from 350 to 700 nm, with an external quantum efficiency of more than 50% between 450 and 550 nm. The photodiodes have a simple layered structure that permits facile integration with planar chip-based systems. To evaluate the suitability of the organic devices as integrated detectors for microscale chemiluminescence, a peroxyoxalate based chemiluminescence reaction (PO-CL) was monitored within a poly(dimethyl-siloxane) (PDMS) microfluidic device. Quantitation of hydrogen peroxide indicated excellent linearity and yielded a detection limit of 10 µM, comparable with previously reported results using micromachined silicon microfluidic chips with integrated silicon photodiodes. The combination of organic photodiodes with PDMS microfluidic chips offers a means of creating compact, sensitive and potentially low-cost microscale CL devices with wide-ranging applications in chemical and biological analysis and clinical diagnostics.
Introduction
The field of microfluidics aims to transform the analytical sciences using techniques developed in the silicon processing industry to engineer miniature devices in which chemical and biological processing can take place under precisely controlled conditions.1,2 Miniaturization of chemical analysis provides several advantages over conventional bulk procedures. These include: (a) reduced analysis times; (b) cost reductions through cheaper fabrication and reduced consumption of reagents; (c) superior control of reaction conditions; (d) enhanced ability to carry out parallel processing; and (e) the ability to perform in-the-field or point-of-care measurements. Typical devices integrate a series of analytical procedures, such as sampling, sample pre-treatment, chemical reactions, analytical separations and detection into a single microchip. The last of these steps is often the most challenging, since only small quantities of analyte are usually present in a microfluidic device, and so high sensitivity detection techniques are needed.One of the preferred methods for analyte detection in microfluidic devices is based around chemiluminescence (CL),3,4 which offers a simple but sensitive means of monitoring low level analyte concentrations. CL reactions typically involve the formation of a metastable reaction intermediate or product in an electronically excited state, which subsequently relaxes to the ground-state with the emission of a photon.5 CL is particularly attractive for portable microfluidic assays, because the CL reaction acts as an internal light-source, thereby lowering instrumental requirements and significantly reducing power consumption and background interference compared to fluorescence assays. Specificity of CL reactions is typically afforded by up-stream enzymatic assays, which generally produce an oxidizing species such as hydrogen peroxide that initiates the CL reaction.3,4 Importantly, the use of CL assays tends to avoid the problematic background emission from interfering compounds or from the microfluidic substrate itself that is frequently encountered in fluorescence detection. CL-based systems have been successfully applied to on-chip electrophoretic separations of metal ions,6,7 immunoassays8 and enzyme assays,9 and consequently there is considerable interest in creating complete analytical devices that incorporate the CL assay and optical detector into a single integrated package.
In previous reports of CL detection in microfluidic environments, the CL signal has generally been detected and quantified using externally mounted photomultiplier tubes (PMTs) and/or microscope collection optics.10–13 Recently, however, Jorgensen et al. reported the use of integrated silicon photodiodes for monitoring luminol-based chemiluminescence reactions in micromachined silicon microfluidic chips.14 They selected hydrogen peroxide as a model compound for quantitation because it is produced by a number of enzymes in the presence of dissolved oxygen and certain analytes such as alcohol, glucose, and cholesterol.15 Using this approach they were able to attain measurable signals down to 10 µM, thus showing that high sensitivity chip-based CL detection could be implemented in a fully integrated microscale format.
The use of silicon photodiodes and micromachined silicon substrates, however, entails relatively high-cost fabrication techniques that preclude the use of such devices in disposable point-of-care applications where low cost is of primary concern. In recent work, therefore, we investigated whether integrated microscale CL could be implemented in a lower cost format using poly(dimethylsiloxane) (PDMS) instead of silicon as the substrate material and organic photodiodes in place of the Si detectors.14 PDMS has good biocompatibility and optical transparency over the visible range,16 and allows for rapid molding-based prototyping and scalable manufacturing at low cost and with high reliability. At the same time, organic devices may be fabricated at low temperature using simple layer-by-layer deposition procedures that are fully compatible with plastic substrates.17–19 The combination of PDMS microfluidic chips with organic photodiodes therefore offers an attractive route to fabricating low-cost diagnostic devices, which incorporate the fluidic channels and the detectors into a single monolithic package.
In our first proof-of-principle studies, we used organic photodiodes based on vacuum deposited bilayers of copper phthalocyanine (CuPc) and fullerene (C60) to detect the emission signal from a peroxyoxalate chemiluminescence (PO-CL) based assay.20 These measurements confirmed the feasibility of using organic devices for detection of the CL signal, but yielded relatively poor detection limits of 1 mM compared to 10 µM reported by Jorgensen et al.14 The modest detection limits were attributable in part to a significant mismatch between the area of the photodiodes (∼16 mm2) and the area of the detection zone on the microfluidic chip (∼2 mm2), which resulted in a high background signal (∼1 nA) due to the dark current.
To improve the limit of detection, it is necessary to substantially reduce the short-circuit dark current in the photodiode. In the work reported here we replaced the 16 mm2 vacuum deposited CuPc/C60 bilayer devices with 1 mm2 solution-processed polymer devices based on 1 : 1 blends by weight of poly(3-hexylthiophene) [P3HT] and [6,6]-phenyl-C61-butyric acid-methylester [PCBM]—a soluble derivative of C60. The P3HT/PCBM devices fabricated in our laboratory typically have very low short-circuit dark current densities of <10−6 mA cm−2, and are consequently a good choice for high sensitivity detection. The 1 mm × 1 mm dimensions of the P3HT:PCBM devices are well matched to the 800 µm × 1 mm detection zone of our microfluidic chips and so minimise the background signal due to the dark current. The thin-film polymer photodiodes, when integrated with PDMS microfluidic chips, provide compact, sensitive and potentially low-cost microscale CL devices with wide-ranging applications in chemical and biological analysis and clinical diagnostics.
Experimental
Microfluidic set-up
The microfluidic chip fabrication was based on standard soft lithography. A master mold was made from negative SU-8 photoresist using standard photolithographic processes.21 The depths of all channels were 800 µm while the respective widths of the inlet and mixing channels were 400 and 800 µm. The inlet channels were 1 cm in length and the mixing channel length was 5.2 cm (Fig. 1A). Using a Sylgard 184 Silicone Elastomer kit (Dow Corning, Coventry, UK), monomer and hardener were mixed at a ratio of 10 : 1 (w/w), degassed for half an hour, poured into a flat molding dish and cured at 95 °C for one hour. To form an enclosed channel, the structured PDMS layer was placed in conformal contact with a 1 mm layer of unstructured PDMS. Intrinsic adhesion between the two layers resulted in a reversible seal and pre-assembly treatment with oxygen plasma yielded permanently bonded PDMS microchips. Fluidic access holes were punched using a blunt syringe needle with 394 µm inner diameter (ID) and 711 µm outer diameter (OD) (BD, Oxford, UK). To yield a more rigid microfluidic test device, a 1 mm thin microscope slide was used as the chip-to-world interface. Holes coinciding with the access holes in the PDMS microfluidic layer were drilled using a 1 mm diameter diamond drill bit. Standard fused silica capillaries with ID 150 µm and OD 367 µm (Composite Metal Services, Hallow, UK) were then inserted and fixed with chemically resistant epoxy to serve as fluidic reservoirs (Araldite 2014, RS Components, Corby, UK). Stinger gas-tight syringes and a Bee syringe pump were employed to hydrodynamically deliver reagents to the microchannels (BAS, West Lafayette, IN, USA). The syringes were connected to 1.6 mm ID high-pressure finger-tights (VWR, Poole, UK) via 762 µm ID PEEK tubing (Supelco, Bellefonte, PA, USA). The outlet of the finger-tights comprised 356 µm ID Teflon tubing (Anachem, Luton, UK) which could be connected to the capillary reservoirs of the PDMS microchip.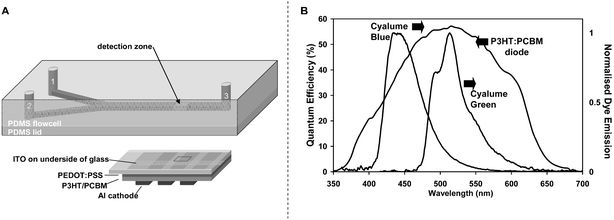 | ||
| Fig. 1 (A) Microchip layout with inlets for premixed CPPO, dye, catalyst (1) and hydrogen peroxide (2), mixing channel and outlet (3). The inlets are 400 µm-wide, 800 µm-deep and 1 cm-long, while the mixing channel is 800 µm-wide, 800 µm-deep and 5.2 cm-long. The active area of the photodiode used for CL detection is 1 mm × 1 mm. The photodiode is located at a position 1 cm downstream from the point-of-confluence of the two inlet streams. (B) The quantum efficiency action spectrum for an ITO/PEDOT:PSS/P3HT:PCBM/Al photodiode and the normalised emission spectra for the two CL dyes used in this work: 9,10-diphenylanthracene (Cyalume blue) and 9,10-bis(phenylethynyl)anthracene (Cyalume green). The emission spectra of both dyes overlap the spectral response of the photodiode. | ||
Organic photodiode fabrication and characterisation
To fabricate the organic photodiode, a 60 nm layer of as-received poly(3,4-ethylene-dioxythiophene):poly-styrene sulfonate (PEDOT:PSS) [Baytron P AI 4083 formulation from H. C. Starck GmbH, Germany] was spin-coated onto a 1 mm ITO-coated glass substrate. The PEDOT:PSS layer was annealed at 140 °C for 30 min in air to remove trapped water vapour, and a 150 nm active layer of regioregular poly(3-hexylthiophene) (P3HT) and 1-(3-methoxycarbonyl)-propyl-1-phenyl-(6,6)C61 (PCBM) was then deposited by spin-coating from a 1 : 1 blend of the two components in dichlorobenzene. The cathode was formed by depositing 200 nm of aluminium through a shadow mask onto the polymer film under vacuum (2 × 10−6 Torr). The active area of the pixel, defined by the spatial overlap of the ITO anode and the Al cathode, was 1 mm × 1 mm. The photodiode was annealed for 30 min at 130 °C in a dry nitrogen atmosphere to improve the blend film morphology18 and was then encapsulated in the same atmosphere by securing a metal can (fitted with a desiccant patch) to the coated side of the glass substrate with a UV-cured adhesive.The photocurrent action spectrum of the polymer device was determined using a 150 W xenon lamp (Bentham Instruments Ltd, Reading, UK), a CM110 monochromator (CVI Technical Optics, Onchan, UK), and a 236 Source-Measure-Unit (SMU) (Keithley, USA). The spectrum was corrected for the intensity of incident light, using a reference spectrum from a calibrated silicon photodiode (UV-818, Newport, UK). The current–voltage characteristics at varying levels of illumination were measured using the Keithley 236 Source-Measure-Unit and the variably attenuated output of a 633 nm laser diode.
Microscale chemiluminescence
The integrated CL device was assembled by attaching the lid of the PDMS microchip to the glass side of the thin-film organic photodiode to form an integrated device in which the organic pixel was aligned with a detection zone on the fluidic chip, located 1 cm downstream of the point-of-confluence of the two inlet streams (see Fig. 1A). To perform the on-chip CL measurements, a stock solution (A) of CL reagent was first prepared by extracting the PO-CL reagents from Cyalume green light-sticks (Omniglow, West Springfield, MA), which contain the active ingredients bis(2-carbopentyloxy-3,5,6-trichlorophenyl) oxalate (CPPO) and the Cyalume green dye 9,10-bis(phenylethynyl)anthracene. 5 mM 4-dimethylaminopyridine (DMAP) was added to serve as a catalyst (Sigma-Aldrich). Test solutions (B) were prepared by diluting 31% aqueous H2O2 stock solution with acetonitrile. Additional measurements were undertaken using the Cyalume blue dye 9,10-diphenylanthracene in place of 9,10-bis(phenylethynyl)anthracene. The two dyes have quantum efficiencies close to unity (0.91 ± 0.08 and 0.85 ± 0.03 for Cyalume green and Cyalume blue respectively) and so are good choices for high sensitivity CL assays.22 The PO-CL reaction uses an indirect emission mechanism in which energy transfer occurs from an excited state intermediate (C2O4) to the dye molecule,23 and hence the observed CL spectrum is determined by the emission spectrum of the selected dye. The solutions A and B were transferred to 0.5 and 1 ml syringes (of equal length and different diameters) respectively, and the syringes were then loaded onto the same drive unit of a Bee syringe pump. In operation, the common drive unit depresses the two syringe plungers at the same rate so, due to the different syringe diameters, the ratio of volumetric injection rates QA : QB was 1 : 2; all flow rates quoted in this paper relate to the total flow rate QT = QA + QB. The short-circuit photocurrent due to the CL emission (referred to hereafter as the CL signal) was measured via spring-loaded probes with a Keithley 236 Source-Measure-Unit.Results and discussion
Fig. 1B shows the short-circuit action spectrum for the 1 mm × 1 mm P3HT:PCBM photodiode. The device has a response over the range 350 to 700 nm, with an external quantum efficiency of more than 50% between 450 and 550 nm. The dark current was 9.44 pA (∼9.44 × 10−7 mA cm−2). These values compare with a peak quantum efficiency of 90% and a dark current density of 4.17 × 10−9 mA cm−2 for a commercially available silicon diode (Newport 818 UV) measured using the same equipment. The shunt-resistance of the organic device—determined at a reverse bias of 10 mV—was equal to 16 kΩ, which implies 1 pA of Johnson noise at a bandwidth of 1 Hz.24Fig. 2A shows, for information, the bias dependence of the photocurrent at varying levels of illumination. The measured IV characteristics are typical of PCBM/P3HT devices reported elsewhere in the literature.25Fig. 2B shows the intensity dependence of the short-circuit photocurrent, which as expected varies linearly with intensity over the full range investigated. All subsequent measurements reported here were obtained under short circuit conditions in order to minimise the dark current and hence background signal. The measured photocurrent in the CL measurements described below is directly proportional to the intensity of the CL emission. The background level before starting the CL reaction (i.e. at zero H2O2 concentration) was 225 pA, which was considerably higher than the measured dark current due to the presence of a small amount of stray light during the CL measurements.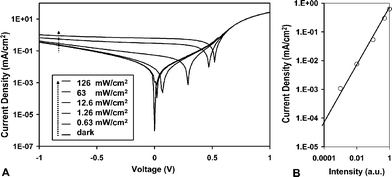 | ||
| Fig. 2 (A) Current–voltage characteristics of the P3HT:PCBM photodiode shown in Fig. 1 under varying levels of 633 nm monochromatic illumination. (B) The intensity dependence of the short-circuit photocurrent for the same device. The signal varies linearly with intensity over the full range investigated. | ||
The chemiluminescence spectra for the blue and green luminescent dyes are shown in Fig. 1B, and are essentially identical to the corresponding photoluminescence spectra for the dyes (not shown) in accordance with the indirect nature of the PO-CL emission mechanism. The emission spectra of the two dyes match reasonably well with the response of the photodiodes and, as noted above, their quantum efficiencies are close to unity so both dyes can be favourably used with the P3HT/PCBM photodiodes. The Cyalume green dye, however, coincides more closely with the peak response of the photodiode and so was used for the measurements reported here.
To initiate the CL reaction, the dye–reagent–catalyst mixture and the H2O2 were pumped hydrodynamically into inlets 1 and 2 respectively (see Fig. 1A) and the signal was detected 1 cm downstream of the point-of-confluence. The CL signal was observed to rise monotonically from zero to a maximum steady-state value over a period of several seconds depending on flow conditions. To determine the optimal flow conditions for later experiments, we first fixed the concentration of H2O2 at 1 mM and investigated the influence of the total flow rate on the measured CL signal. The variation of the CL signal in response to changes in the flow rate is shown in Fig. 3, where the black arrows denote the points in time at which the flow rate was changed. In each case, steady-state was reached within ∼20 s of adjusting the flow rate. The data shows the progression in CL signal as the total flow rate was sequentially changed from 0 to 150 to 75 to 30 to 0 to 150 and back to 0 µl min−1. A total flow rate of 150 µl min−1 resulted in the strongest CL signal with excellent reproducibility being obtained between the second and the penultimate flow rates (both 150 µL min−1). The slight fluctuations in the steady-state CL signal are due to small gas bubbles generated in the early stages of the reaction that scatter the emitted light.
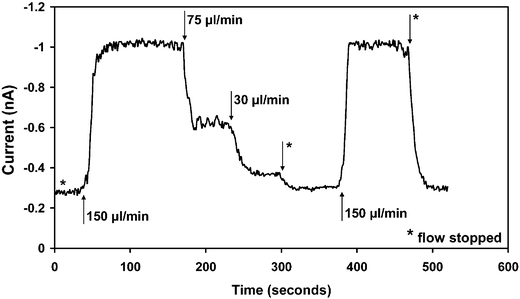 | ||
| Fig. 3 Transient CL signal for the on-chip mixing of 1 mM hydrogen peroxide with CPPO, 9,10-bis(phenylethynyl)anthracene and 5 mM DMAP. The data show the progression in CL signal as the total flow rate was sequentially changed from 0 to 150 to 75 to 30 to 0 to 150 and back to 0 µL min−1. The arrows indicate the times at which the flow rate was adjusted, and the asterisks represent 0 µL min−1, i.e. stopped flow. The most stable CL signal was obtained at a total flow rate of 75 µL min−1, and this flow rate was therefore selected for subsequent measurements. | ||
The intensity of the measured CL signal is dependent on the mixing dynamics within the channel: at the optimal flow rate, substantial mixing of the reagents has already occurred by the time they reach the detector, leading to a strong CL signal; at higher flow rates, the reagents will not have mixed adequately by the time they reach the detection zone, leading to a reduced CL signal; and, at excessively slow flow rates, the reaction is partially over by the time the detection zone is reached, leading again to a reduced signal. For the Cyalume reagents selected, the catalysed CL reaction proceeds quickly so the strongest emission is obtained at the highest flow rate of 150 µL min−1. In principle, the CL emission strength could be enhanced by raising the flow rate further still. However, at flow rates above 100 µL min−1, the system was prone to occasional periods of excessive effervescence, resulting in an intermittently unstable signal (not shown). The effervescence is most severe in the earliest stages of the reaction and for reasons of reliability it is preferable to use lower flow rates, which allow the reaction to ‘settle down’ before the detection zone is reached, resulting in a weaker but significantly more stable signal. In practice, a flow rate of 75 µL min−1 was found to provide a good compromise between long term signal stability (over a time-scale of several minutes) and absolute intensity.
The total flow rate was fixed at 75 µL min−1 to investigate the influence of H2O2 concentration on the intensity of the CL signal. H2O2 solutions of sequentially increasing concentration were pumped into inlet 2, and the CL reagent with catalyst and dye were pumped into inlet 1. The transient response of the CL signal at each of the H2O2 concentrations is shown in Fig. 4A. In each case steady-state is reached within approximately 10 s, which is important for real-world applications that require fast sample-to-answer times. At concentrations above 100 µM there is a slight overshoot in the signal before steady-state is reached which may relate to a change in the mixing dynamics within the chip. The signal nonetheless reaches steady-state within a short period of time and the overshoot does not adversely affect analytical performance. The steady-state CL signal is plotted against hydrogen peroxide concentration in Fig. 4B, and excellent linearity is obtained in the range 10 µM to 1 mM with a correlation coefficient, R, of 0.997. (A slight deviation from linearity was observed at higher H2O2 concentrations due presumably to an insufficient amount of dye or reagent to react with hydrogen peroxide.) The detection limit for the integrated chip/detector was <10 µM, which represents a 100-fold enhancement compared to the data we reported previously.20 Significantly, the sensitivity of the integrated CL devices described here is equivalent to that of the devices reported by Jorgensen et al. using integrated silicon photodiodes.14
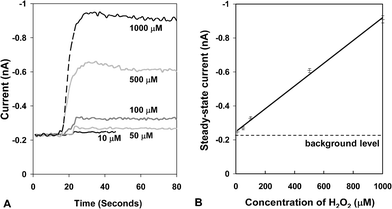 | ||
| Fig. 4 (A) Transient CL signals for the on-chip mixing of CPPO, 9,10-bis(phenylethynyl)anthracene and 5 mM DMAP with various concentrations of hydrogen peroxide at a total applied flow rate of 75 µL min−1. (B) The steady-state CL signal as a function of H2O2 concentration extracted from the data in (A). The error bars are deduced from the fluctuating (steady-state) transient signals and correspond to three standard deviations either side of the mean value. | ||
Conclusion
The limit of detection for our devices is determined primarily by the background signal on the organic photodiodes, which could be further reduced by screening the devices more effectively from stray light and reducing the short-circuit dark currents through improved device fabrication. In particular, reducing the surface roughness of the ITO substrate should reduce the probability of pin-hole formation in the polymer layers and thus reduce the dark current due to shunt conductance. In addition, very recent studies by Kim et al. have indicated that an active layer thickness of 175 nm results in a slightly improved quantum efficiency compared with the devices reported here (∼70% versus ∼60%).26 These devices should exhibit significantly lower short-circuit dark currents due to the reduced tendency for filament formation in thick films. Changes to the CL chemistry may also enable appreciable increases in emission intensity since the Cyalume dyes we use are primarily optimised for duration of emission and, in the current context, a short-lived high intensity emission is preferable.27 We anticipate that taken together these changes may enable a further 100 to 1000-fold improvement in the limit-of-detection. The current 10 µM limit-of-detection is already sufficient for many diagnostic applications including the determination of alcohol, glucose, and cholesterol levels in blood.4 Further improvements to the 100 nM level would provide sufficient sensitivity for applications such as low level cancer marker detection28 and pharmakinetics.29 Finally, we note that these initial studies were conducted in a lab environment using standard support instrumentation that is ill-suited to miniaturized low-cost applications. In ongoing studies, we are evaluating the use of on-chip reagent storage methods27 and passive capillarity-based fluid delivery schemes that remove the need for an external syringe pump. Furthermore we are investigating the use of integrated electronics for read-out and display of the photodiode current. The resultant devices would offer a powerful low-cost solution for chemical and biological analysis with potentially wide-ranging applications for in-the-field analysis and point-of-care diagnostics.Acknowledgements
The authors acknowledge support from the UK Biotechnology and Biological Sciences Research Council through its Small Business Research Initiative (grant 147/SBRI 19689), and are grateful to Merck Chemicals Ltd for the provision of P3HT and to LG Philips LCD for provision of substrates and desiccants. The authors are also grateful to Mr Sung Joon Bae for useful discussions. The Molecular Vision authors are grateful to Acrongenomics for financial assistance towards this work.References
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1, 244 CrossRef.
- D. R. Reyes, D. Iossifidis, P. A. Auroux and A. Manz, Anal. Chem., 2002, 74, 2623 CrossRef CAS.
- P. Fletcher, K. N. Andrew, A. C. Calokerinos, S. Forbes and P. J. Worsfold, Luminescence, 2001, 16, 1 CrossRef CAS.
- Z. Y. Zhang, S. C. Zhang and X. R. Zhang, Anal. Chim. Acta, 2005, 541, 37 CrossRef CAS.
- A. M. Garcia-Campana, W. R. G. Baeyens, X. R. Zhang, E. Smet, G. Van der Weken, K. Nakashima and A. C. Calokerinos, Biomed. Chromatogr., 2000, 14, 166 CrossRef CAS.
- K. Tsukagoshi, M. Hashimoto, R. Nakajima and A. Arai, Anal. Sci., 2000, 16, 1111 CAS.
- B. F. Liu, M. Ozaki, Y. Utsumi, T. Hattori and S. Terabe, Anal. Chem., 2003, 75, 36 CrossRef CAS.
- J. Yakovleva, R. Davidsson, M. Bengtsson, T. Laurell and J. Emneus, Biosens. Bioelectron., 2003, 19, 21 CrossRef CAS.
- T. Richter, L. L. Shultz-Lockyear, R. D. Oleschuk, U. Bilitewski and D. J. Harrison, Sens. Actuators, B, 2002, 81, 369 CrossRef.
- S. D. Mangru and D. J. Harrison, Electrophoresis, 1998, 19, 2301 CAS.
- T. Kamidate, T. Kaide, H. Tani, E. Makino and T. Shibata, Luminescence, 2001, 16, 337 CrossRef CAS.
- R. G. Su, J. M. Lin, F. Qu, Z. F. Chen, Y. H. Gao and M. Yamada, Anal. Chim. Acta, 2004, 508, 11 CrossRef CAS.
- J. G. Lv and Z. J. Zhang, Anal. Lett., 2004, 37, 1401 CrossRef.
- A. M. Jorgensen, K. B. Mogensen, J. P. Kutter and O. Geschke, Sens. Actuators, B, 2003, 90, 15 CrossRef.
- X. R. Zhang, W. R. G. Baeyens, A. M. Garcia-Campana and J. Ouyang, Trends Anal. Chem., 1999, 18, 384 CrossRef CAS.
- D. Qin, Y. N. Xia, A. J. Black and G. M. Whitesides, J. Vac. Sci. Technol., B, 1998, 16, 98 CrossRef CAS.
- B. Y. Ouyang, C. W. Chi, F. C. Chen, Q. F. Xi and Y. Yang, Adv. Funct. Mater., 2005, 15, 203 CrossRef CAS.
- G. Yu, K. Pakbaz and A. J. Heeger, J. Electron. Mater., 1994, 23, 925 Search PubMed.
- J. Huang, A. J. deMello, D. D. C. Bradley and J. C. deMello, Phys. Chem. Chem. Phys., 2006, submitted Search PubMed.
- O. Hofmann, P. Miller, P. Sullivan, T. S. Jones, J. C. deMello, D. D. C. Bradley and A. J. deMello, Sens. Actuators, B, 2005, 106, 878 CrossRef.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974 CrossRef CAS.
- P. J. Hanhela and D. B. Paul, Aust. J. Chem., 1984, 37, 553 CAS.
- P. Lechtken and N. J. Turro, Mol. Photochem., 1974, 6, 95 Search PubMed.
- http://sales.hamamatsu/assets/applications/SSD/photodiode_technical_information.pdf. .
- M. Reyes-Reyes, K. Kim and D. L. Carroll, Appl. Phys. Lett., 2005, 87.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C. S. Ha and M. Ree, Nat. Mater., 2006, 5, 197 CrossRef CAS.
- M. Stigbrand, E. Ponten and K. Irgum, Anal. Chem., 1994, 66, 1766 CrossRef CAS.
- K. Tsukagoshi, N. Jinno and R. Nakajima, Anal. Chem., 2005, 77, 1684 CrossRef CAS.
- Z. J. Zhang, D. Y. He, W. Liu and Y. Lv, Luminescence, 2005, 20, 377 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |