A microfluidic bioreactor for increased active retrovirus output†
Halong N.
Vu‡
,
Yawen
Li‡
,
Monica
Casali
,
Daniel
Irimia
,
Zaki
Megeed
and
Martin L.
Yarmush
*
Center for Engineering in Medicine and Department of Surgery, BioMEMS Resource Center, Massachusetts General Hospital, Shriners Hospital for Children, and Harvard Medical School, 51 Blossom Street, Rm. 406, Boston, MA 02114, USA. E-mail: ireis@sbi.org; Fax: +1-(617)-371-4882; Tel: +1-(617)-371-4950
First published on 30th October 2007
Abstract
Retroviruses are one of the most commonly used vectors in ongoing gene therapy clinical trials. To evaluate and advance virus production on the microscale platform, we have created a novel microfluidic bioreactor for continuous retrovirus production. We investigated the growth kinetics of a retroviral packaging cell line in microfluidic bioreactors for several compartment sizes, and packaging cells perfused in the microdevices showed similar growth kinetics to those cultured in conventional static conditions. To evaluate the efficiency of retrovirus production, virus titers from the microdevices were compared to those obtained from static tissue culture. When retrovirus production and collection were maintained at 37 °C, virus production levels were comparable for the microdevices and static tissue culture conditions. However, immediate cold storage downstream of the packaging cells in the microdevices resulted in 1.4- to 3.7-fold greater active virus production levels with the microdevices compared to the conventional static conditions over a 5 day period. Lastly, the use of microfluidics for virus production provides a continuous supply of virus supernatant for immediate infection of target cells or for preservation and storage. Such devices will be valuable for the optimization of production and evaluation of retroviruses and other viral vectors for gene therapy applications.
Introduction
Retroviruses remain one of the most commonly used vectors in gene therapy clinical trials to date, comprising nearly 25% of all ongoing trials.1 Due to its ability to deliver genes permanently into target cells, a retroviral vector was used to treat patients with advanced melanoma in the first human gene therapy trial, in 1989.2 Retroviruses have also played critical roles in the success of more recent gene therapy clinical trials.3–5 However, the short half-life of retroviruses, ranging from 4 to 9 h at 37 °C,6–8 limits the maximum titers achievable for these vectors. As a result, the challenges of retrovirus production and purification represent two major technical barriers to their clinical progress.8–11 Retroviral vectors are produced by packaging cells that are typically fibroblast derivatives containing DNA plasmids to express specific viral gene products.12,13 With reports on the production of retroviral vectors indicating a strong dependence of viral titer on culturing conditions,8,14 it would be desirable to have a high throughput platform for optimizing virus production and treatment conditions, which is otherwise labor-intensive and time-consuming using conventional tissue culture methods.Microfabricated devices and microfluidics are expected to have an immense impact in the near future, with researchers exploring their applications in broad areas of biology and medicine.15–17 Compared to conventional cell culture methods, microsystems offer unique advantages of smaller reagent volumes, shorter retention times, and the possibility of parallel processing and analysis. The micron scale components enable a higher degree of control over environmental conditions, which may also enable one to provide a more physiologically relevant platform that mimics the in vivo cellular microenvironment.18 In light of these advantages, we aimed to adapt retrovirus production to the microscale platform and assess the feasibility of using microsystems for identifying improved culturing conditions. Previously, microfluidics has been applied to generate gradients of virus in solutions,19 dielectrophoretic capture and imaging of viral particles on microelectrodes,20 and cell micropatterning has been used to observe virus propagation and infection in cell arrays.21
We hypothesized that a microfluidic bioreactor would enable the seeding of packaging cells at a very high surface to volume ratio and allow us to attain better-controlled microenvironments for rapid and reproducible evaluation of various culturing conditions, not possible with static methods, to improve the production of retroviral vectors. Further, the use of a microfluidic bioreactor for virus production would generate a continuous stream of freshly-produced virus-containing medium at the outlet for immediate use, e.g., transduction of target cells, or for preservation and storage. Here, a novel microfluidic bioreactor was designed and fabricated for continuous production of retroviral vectors in a commercially available packaging cell line. The effects of channel height, shear stress, and perfusion on the packaging cell growth and viral activity were investigated, and these results were compared to cell growth and virus production from conventional static cell culture conditions.
Materials and methods
Device design and fabrication
A microfluidic device with 8 parallel channels (per channel: 30 × 2.5 mm) was designed for the study of virus production (Fig. 1A). Three different channel heights were tested (50, 100, and 150 µm) to consider the effect of compartment size and shear stress on cell growth and virus production. The devices were fabricated using the polydimethylsiloxane (PDMS) soft lithography technique.22 Briefly, the masks were drawn using AutoCAD 2002 and printed on transparencies with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dpi resolution (Fineline Imaging, Colorado Springs, CO, USA), and masters were created using a negative resist SU-8 (Microchem Inc., Newton, MA, USA) on silicon wafers using UV photolithography. PDMS devices were molded from the masters using the two-part Sylgard 184 silicone elastomer (Dow Corning, Midland, MI, USA). Inlets and outlets were punched through the devices using a sharpened needle (NE251PL, Small Parts Inc., Miami Lakes, FL, USA). The PDMS devices were treated with oxygen plasma (50 W, 2% O2, 25 s), bonded to glass slides, and heated (100 °C, 10 min) to achieve permanent bonding. Tygon tubings (TGY010, Small Parts Inc., Miami Lakes, FL, USA) were inserted in the inlet and outlet holes.
000 dpi resolution (Fineline Imaging, Colorado Springs, CO, USA), and masters were created using a negative resist SU-8 (Microchem Inc., Newton, MA, USA) on silicon wafers using UV photolithography. PDMS devices were molded from the masters using the two-part Sylgard 184 silicone elastomer (Dow Corning, Midland, MI, USA). Inlets and outlets were punched through the devices using a sharpened needle (NE251PL, Small Parts Inc., Miami Lakes, FL, USA). The PDMS devices were treated with oxygen plasma (50 W, 2% O2, 25 s), bonded to glass slides, and heated (100 °C, 10 min) to achieve permanent bonding. Tygon tubings (TGY010, Small Parts Inc., Miami Lakes, FL, USA) were inserted in the inlet and outlet holes.
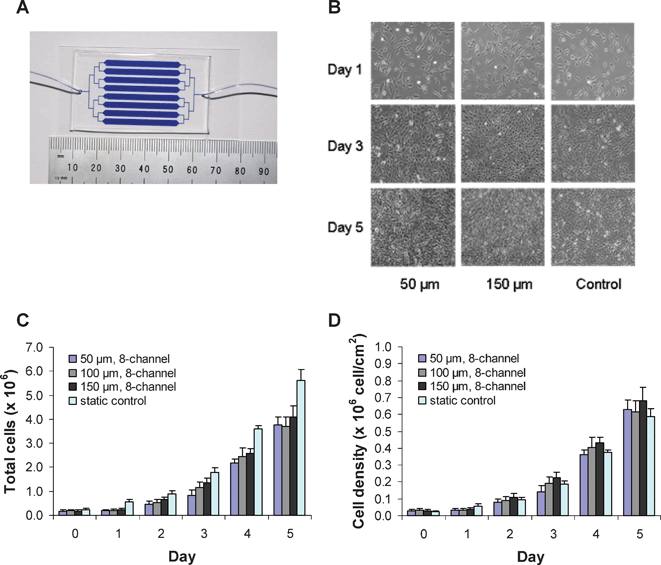 | ||
| Fig. 1 A microfluidic bioreactor for continuous virus production. (A) The microfluidic device with 8 parallel channels was formed by molding PDMS against an SU-8 master. Microdevices for continuous virus production were made with 50, 100, and 150 µm channel heights. The device shown was filled with blue dye for visualization. (B) Phase contrast images of PT67 packaging cells growing in 50 and 150 µm channel height microdevices under 2 µl min–1 perfusion over 5 days, and images on the right are from the 6-well static control conditions. (C) Total cell number and (D) cell density of the packaging cells growing in the microdevices and in the static control 6-well plate (n ≥ 3; error bars represent standard deviation). | ||
Device seeding and cell culture
Devices were UV sterilized overnight, and coated with fibronectin (25 µg ml–1; Sigma, St. Louis, MO, USA) for 2 h at 37 °C. Channels were then seeded with PT67-GFP packaging cells (Clontech Laboratories Inc., Mountain View, CA, USA), producing replication-deficient murine leukemia virus (MLV) retroviruses harboring the green fluorescent protein (GFP) reporter gene. The cell culture medium was Dulbecco's Modified Eagle medium (DMEM; Gibco BRL, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone Labs Inc., Logan, UT, USA), 100 U ml–1penicillin and 100 µg ml–1streptomycin (Gibco BRL), 1% bovine serum albumin (BSA, Sigma) and 25 mM HEPES (Sigma).Different cell densities were used to seed devices with different channel heights, in order to obtain the same number of cells attached on the device surface. Namely, 2 × 106cells ml–1 were used to seed devices with 50 µm channel height, 1 × 106cells ml–1 for devices with 100 µm channel height, and 0.67 × 106cells ml–1 for devices with 150 µm channel height. This led to the same degree of confluency after cell attachment. The seeded devices were incubated overnight at 37 °C, in 10% CO2 atmosphere, and the perfusion initiated the following morning. Continuous flow of medium was controlled by a multichannel syringe pump (Harvard Apparatus, Holliston, MA, USA) set at 2 µl min–1 for all microdevice experiments. Devices were monitored daily for cell growth by phase contrast microscopy. For the static control, 2 ml of 5 × 104cells ml–1 were used to seed the PT67-GFP packaging cells in a conventional 6-well tissue culture plate (Dow Corning). The virus-containing media were collected daily, and virus samples were frozen and stored at –80 °C until use. The cell number was determined by treating the cells with trypsin (Gibco, BRL), redispersing the cells in 1 ml medium, and counting the cells with a hemocytometer. Coating the tissue culture plates with fibronectin did not affect cell growth or virus titer compared to the untreated 6-well plates.
GFP transduction assay
NIH 3T3 cells (CRL-1658; ATCC, Manassas, VA, USA) were used to evaluate the transduction efficiency of the PT67-GFP virus samples from the microdevices and static tissue culture conditions. Briefly, thirty thousand 3T3 cells in 1 ml medium per well were plated in a 12-well flat bottom tissue culture dish (Dow Corning). The next day, the medium was replaced with 1 ml fresh medium containing 100 µl viral solution and 8 µg ml–1 polybrene (Sigma). After overnight virus exposure, the medium was replaced with fresh virus-free medium and cells were cultured for another day. Infected cells expressing GFP were observed by a Zeiss Axiovert 200 M inverted microscope. Quantitative evaluation of virus infection was performed by flow cytometry for GFP on a Coulter EPICS ALTRA flow cytometer (Beckman Coulter, Miami, FL, USA).Results and discussion
Cell growth and cell morphology
The 50, 100, and 150 µm channel height microdevices and the 6-well static culture plate were seeded to obtain a similar level of confluency after cell attachment to the surface. PT67-GFP packaging cells were ∼40% confluent the day after seeding, in both the microdevices and the 6-well plates. After 5 days of perfusion, the cells in the microdevices and in the static culture reached nearly 100% confluency (Fig. 1B). The total number of cells was determined at each day in the microdevices and static cultures (Fig. 1C). The doubling time of the packaging cells was approximately 24 h for all the configurations tested. However, little growth was observed for cells in the microdevices during the first 24 h. This may reflect the time needed for the cells to adapt to the microfluidic device or may be related to nutrient transport issues. While the total number of packaging cells was highest in the 6-well static tissue culture plate throughout the 5 day period, the cell densities were similar in the 6-well plate and microfabricated flowing systems (Fig. 1D). The data show that the rate of proliferation of packaging cells in the microdevices and in the 6-well plate were comparable, indicating that packaging cell growth was not adversely affected by the flow or culture in the microfluidic device. The cell morphology was also similar in the microdevice and static culture conditions, as depicted in Fig. 1B.Cell proliferation appeared to be independent of the microdevice channel height when the flow rate was held constant at 2 µl min–1. With the Navier–Stokes equation for Newtonian fluid flow between parallel plates (τ = 6ηQ/h2b; for shear stress τ, dynamic viscosity η, flow rate Q, channel height h, and channel width b), we estimated the shear stress on the packaging cells in the 50, 100, and 150 µm height microdevices to be 0.03, 0.07, and 0.003 dyn cm–2, respectively. These values are much lower than the reported values of 50 to 60 dyn cm–2 for 3T3 fibroblasts to detach from glass substrates.23,24 Thus, the PT67-GFP packaging cell morphology and proliferation were insensitive to the change in shear stress over the range of shear stresses investigated in our study.
Virus production in microfluidic devices at 37 °C
After establishing that packaging cell proliferation and morphology were maintained in the microdevices, we quantified the viral titer and production rates in the microfluidic devices and compared it to the 6-well plate. Cells were cultured in the microdevices at 37 °C, and virus-containing supernatant was continuously harvested for 24 h collection periods and then frozen and stored at –80 °C. With a constant flow rate of 2 µl min–1, the total volume of viral supernatant collected from each microdevice per 24 h period was 2.9 ml. The viral titers from the 50, 100, and 150 µm height microdevices were ∼15 to 46% of the titers from the 6-well plates over the 5 day period (Fig. 2A). Viral titers for the microdevices plateaued at ∼3 × 105IU ml–1 (IU = infectious units), while titers in the 6-well plates plateaued at ∼1.3 × 106IU ml–1. Viral titers were independent of the shear stress in the microdevices, which is consistent with the packaging cell proliferation kinetics described above and in Fig. 1.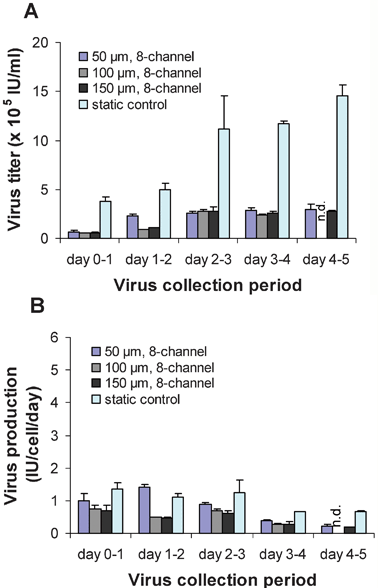 | ||
| Fig. 2 Virus production and collection at 37 °C. (A) Virus titers and (B) virus production for the 50, 100, and 150 µm height microdevices and the 6-well static control for 24 h collection periods over 5 days (n = 3; error bars represent standard deviation; n.d., not done). | ||
When calculating viral production rates, we take into account the cell number and total volume of virus collected to obtain the number of infectious viral particles produced per cell per day (IU cell–1 day–1). The higher virus titer in the static cultures is due to both the differences in the cell number and the fluid volume. The static cultures generate 2 ml of virus supernatant per day, while the microfluidic devices generate 2.9 ml. Additionally, the microdevices contain fewer virus producer cells than the static cultures (Fig. 1C). After adjusting for these two factors (fluid volume and cell number), the viral production rates in the microdevice were closer to those in the 6-well plates (Fig. 2B). Viral production levels averaged 1.0 IU cell–1 day–1 for the static 6-well plate and 0.6 IU cell–1 day–1 for the microdevices over the 5 day period. The somewhat lower production rates observed in the case of the microdevices may be due in part to the adsorption of viral particles to the walls of the channels, as the hydrophobicity of PDMS has been reported to cause protein adsorption.15,25 In a preliminary study, we observed that producing retrovirus in a microdevice with perfusion medium consisting of DMEM with 10% FBS, penicillin and streptomycin resulted in a significant reduction of bioactive virus or viral proteins, due to adsorption to the PDMS channels. Adding 1% BSA to the perfusion medium was shown to reduce viral adsorption to the channels by more than 60% (data not shown). Even though BSA was found to inhibit adsorption, some degree of viral protein adsorption to the PDMS channels still occurred. This remaining adsorption may explain the difference in virus production rate compared to the static cell cultures.
Virus production in microfluidic devices with cold storage compartment
A primary benefit of utilizing microdevices and microfluidics is that it allows precise control over the inlet and outlet flow streams, and compartmentalization of on-chip and off-chip processes. Retroviruses are known to degrade rapidly at 37 °C,6–8 and it has been reported that low temperature storage can slow viral degradation kinetics and hence increase the half-life of the virus.7,26 Consequently, we sought to reduce virus degradation and improve viral production efficiency by connecting the outlet stream from the microdevice to a low temperature preservation compartment, for immediate cold storage. Packaging cells were cultured as previously described and the virus-containing media was continuously transported to a downstream cold storage area (10–0 °C) throughout the 24 h collection period (Fig. 3A). This arrangement, which is not readily feasible with conventional tissue culture methods, was found to increase the viral titer considerably, to levels comparable to or greater than titers from the 6-well plates (Fig. 3B). The conventional tissue culture virus samples were harvested after 24 h intervals as before. Viral titers for microdevices with immediate cold storage plateaued at ∼1.6 × 106IU ml–1 after 3 days of perfusion, representing a 5-fold increase over titers from microdevices without cold storage. Furthermore, viral production levels for the microdevices with cold storage (averaging 2.5 IU cell–1 day–1) were 1.4- to 3.7-fold greater compared to virus production in the static conditions over the 5 days (Fig. 3C). Thus, with the use of microdevices and control over the flow, the active virus production efficiency of the PT67 packaging cells was markedly improved by incorporating a low temperature collection compartment to slow virus degradation.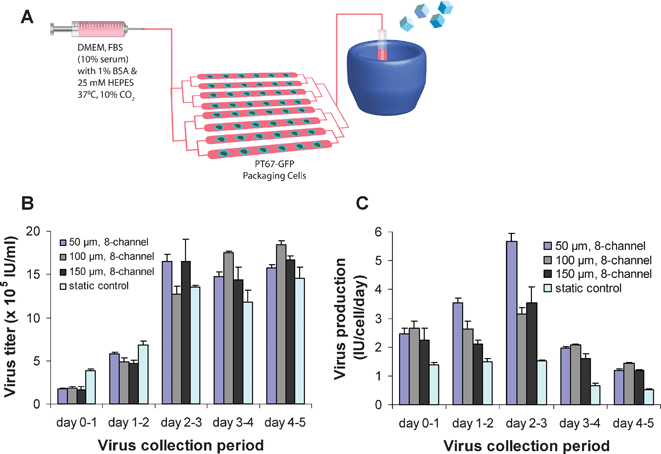 | ||
| Fig. 3 Virus production with downstream cold storage compartment. (A) Schematic of experimental setup for the incorporation of low temperature preservation for virus sample collection with the microdevices. (B) Virus titers and (C) virus production for the 50, 100, and 150 µm height microdevices with cold storage and the 6-well static control for 24 h collection periods over 5 days (n = 3; error bars represent standard deviation). | ||
Future directions
To facilitate our ongoing studies, aimed at incorporating microfluidic virus production and transduction processes for more useful on-chip applications, we performed a simple experiment to detect the transduction of target cells in a microdevice. A 150 µm channel height microdevice was seeded with 3T3 target cells and incubated overnight. The following day, medium containing virus was introduced into the microdevice at a constant flow rate of 2 µl min–1. Two days later, infection of 3T3 target cells could be easily observed under a fluorescence microscope (Fig. 4). With these tools at hand, it is now feasible to integrate virus production with multiple transduction chambers for further optimization of virus production or for the study of virus degradation or maturation with newly produced virus samples generated continuously from the microfluidic bioreactor (Fig. 4). We also envision creating a mutant virus population in the microdevice production compartment to efficiently screen the mutants for infection of numerous cell types at once, in multiple downstream transduction compartments. Moreover, viral transduction of cells in microdevices may be used to more aptly study and model viral targeting or infection dynamics for viruses in flowing liquids, such as viruses in the bloodstream. Microvalves27 can be introduced to provide continuous perfusion and precise downstream control of the viral medium. It would be advantageous to design the system to mimic natural conditions for examining virus adsorption, binding, penetration, and transduction in diverse physiological settings.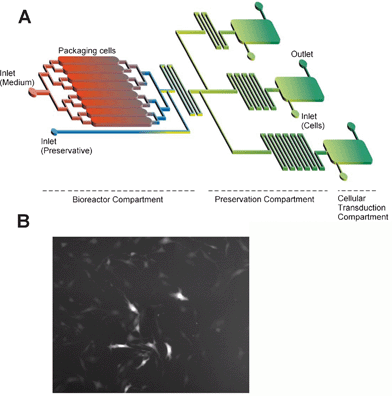 | ||
| Fig. 4 Transduction of target cells in the microdevice and future prospects. (A) Design of an integrated production and analysis system for optimization of viral production and transduction processes, and (B) fluorescence image of 3T3 fibroblasts in a microchannel infected by viruses flowing in the perfusion medium. | ||
Conclusions
In this study, we have successfully demonstrated the production of retroviruses in a microdevice with the commercially available PT67 packaging cell line. The similar growth kinetics of packaging cells in the microdevice and in the static culture has implications for further optimization of the culturing and treatment conditions for retrovirus production. In addition, the microfluidic bioreactor makes available a continuous outlet stream of virus supernatant for immediate storage/preservation or for infection of target cells. This work also provides guidance for the development of an integrated cell culture and analysis system, which may be useful for the production and evaluation of bioactive retroviruses and other viral vectors for gene therapy applications.Acknowledgements
We are thankful for the constructive feedback and technical support with the microfabrication procedures from Octavio Hurtado and Kevin King. The authors would like to thank Don Poulsen and Z Kelley for technical support. We would like to acknowledge the NIH BioMEMS Resource Center Grant P41 EB002503 and the Shriners Hospital.References
- J. Gene Med., 2007, http://www.wiley.co.uk/genetherapy/clinical/,, accessed July 2007 Search PubMed.
- S. A. Rosenberg, P. Aebersold, K. Cornetta, A. Kasid, R. A. Morgan, R. Moen, E. M. Karson, M. T. Lotze, J. C. Yang and S. L. Topalian, N. Engl. J. Med., 1990, 323, 570–578 CAS.
- M. Cavazzana-Calvo, S. Hacein-Bey, G. de Saint Basile, F. Gross, E. Yvon, P. Nusbaum, F. Selz, C. Hue, S. Certain, J. L. Casanova, P. Bousso, F. L. Deist and A. Fischer, Science, 2000, 288, 669–672 CrossRef.
- A. Aiuti, S. Slavin, M. Aker, F. Ficara, S. Deola, A. Mortellaro, S. Morecki, G. Andolfi, A. Tabucchi, F. Carlucci, E. Marinello, F. Cattaneo, S. Vai, P. Servida, R. Miniero, M. G. Roncarolo and C. Bordignon, Science, 2002, 296, 2410–2413 CrossRef CAS.
- M. G. Ott, M. Schmidt, K. Schwarzwaelder, S. Stein, U. Siler, U. Koehl, H. Glimm, K. Kuhlcke, A. Schilz, H. Kunkel, S. Naundorf, A. Brinkmann, A. Deichmann, M. Fischer, C. Ball, I. Pilz, C. Dunbar, Y. Du, N. A. Jenkins, N. G. Copeland, U. Luthi, M. Hassan, A. J. Thrasher, D. Hoelzer, C. von Kalle, R. Seger and M. Grez, Nat. Med., 2006, 12, 401–409 CrossRef CAS.
- S. T. Andreadis, D. Brott, A. O. Fuller and B. O. Palsson, J. Virol., 1997, 71, 7541–7548 CAS.
- J. M. Le Doux, H. E. Davis, J. R. Morgan and M. L. Yarmush, Biotechnol. Bioeng., 1999, 63, 654–662 CrossRef.
- O. W. Merten, J. Gene Med., 2004, 6, S105–124 CrossRef CAS.
- S. K. Powell, M. A. Kaloss, A. Pinkstaff, R. McKee, I. Burimski, M. Pensiero, E. Otto, W. P. Stemmer and N. W. Soong, Nat. Biotechnol., 2000, 18, 1279–1282 CrossRef CAS.
- P. L. Sheridan, M. Bodner, A. Lynn, T. K. Phuong, N. J. DePolo, D. J. de la Vega, Jr., J. O'Dea, K. Nguyen, J. E. McCormack, D. A. Driver, K. Townsend, C. E. Ibanez, N. C. Sajjadi, J. S. Greengard, M. D. Moore, J. Respess, S. M. Chang, T. W. Dubensky, Jr., D. J. Jolly and S. L. Sauter, Mol. Ther., 2000, 2, 262–275 CrossRef CAS.
- S. T. Andreadis, C. M. Roth, J. M. Le Doux, J. R. Morgan and M. L. Yarmush, Biotechnol. Prog., 1999, 15, 1–11 CrossRef CAS.
- J. M. Le Doux, J. R. Morgan, R. G. Snow and M. L. Yarmush, J. Virol., 1996, 70, 6468–6473.
- K. M. Kurian, C. J. Watson and A. H. Wyllie, Mol. Pathol., 2000, 53, 173–176 CrossRef CAS.
- O. W. Merten, P. E. Cruz, C. Rochette, C. Geny-Fiamma, C. Bouquet, D. Goncalves, O. Danos and M. J. Carrondo, Biotechnol. Prog., 2001, 17, 326–335 CrossRef CAS.
- P. A. Auroux, D. Iossifidis, D. R. Reyes and A. Manz, Anal. Chem., 2002, 74, 2637–2652 CrossRef CAS.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS.
- G. M. Walker, H. C. Zeringue and D. J. Beebe, Lab Chip, 2004, 4, 91–97 RSC.
- G. M. Walker, M. S. Ozers and D. J. Beebe, Sens. Actuators, B, 2004, 98, 347–355 CrossRef.
- D. Akin, H. Li and R. Bashir, Nano Lett., 2004, 4, 257–259 CrossRef CAS.
- E. E. Endler, K. A. Duca, P. F. Nealey, G. M. Whitesides and J. Yin, Biotechnol. Bioeng., 2003, 81, 719–725 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS.
- G. A. Truskey and J. S. Pirone, J. Biomed. Mater. Res, .1990, 24, 1333–1353 Search PubMed.
- A. S. Goldstein and P. A. DiMilla, J. Biomed. Mater. Res., 2002, 59, 665–675 CrossRef CAS.
- Y. S. Shin, K. Cho, S. H. Lim, S. Chung, S.-J. Park, C. Chung, D.-C. Han and J. K. Chang, J. Micromech. Microeng., 2003, 768–774 Search PubMed.
- H. Kotani and P. B. Newton, Hum. Gene Ther., 1994, 5, 19–28 CrossRef CAS.
- D. Irimia and M. Toner, Lab Chip, 2006, 6, 345–352 RSC.
Footnotes |
| † The HTML version of this article has been enhanced with colour images. |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2008 |