Employ a bisthienothiophene linker to construct an organic chromophore for efficient and stable dye-sensitized solar cells†
Guangliang
Zhang
a,
Yu
Bai
a,
Renzhi
Li
a,
Dong
Shi
a,
Sophie
Wenger
b,
Shaik M.
Zakeeruddin
b,
Michael
Grätzel
b and
Peng
Wang
*a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: peng.wang@ciac.jl.cn; Fax: +86 431 852 629 53; Tel: +86 431 852 629 52
bLaboratory for Photonics and Interfaces, Swiss Federal Institute of Technology, CH1015, Lausanne, Switzerland
First published on 27th November 2008
Abstract
We employed the bisthienothiophene conjugated linker along with a hydrophobic triphenylamine electron-donor and a hydrophilic cyanoacrylic acid electron-acceptor to construct a high molar extinction coefficient organic photosensitizer, exhibiting a power conversion efficiency of 8.0% measured under irradiation of air mass 1.5 global (AM 1.5G) full sunlight.
Broader contextAt the moment a major drawback of the dye-sensitized solar cell (DSC) technology is the usage of volatile electrolytes to achieve high efficiency. This hurdle has precluded large-scale outdoor application and integration into flexible cells. Amongst various efforts to address this issue, the use of room-temperature ionic liquid electrolytes has proved to be the most successful strategy by far. However, with a low fluidity ionic liquid electrolyte the charge collection yield in DSCs decreases due to the shortened electron diffusion length. Enhancing the optical absorption coefficient of a stained mesoporous film to allow for the use of a thin photoactive layer can counter this effect. In view of the limited ruthenium resource, metal-free organic dyes with a high molar extinction coefficient have attracted considerable attention in the recent years owing to their low-cost and good flexibility in molecular tailoring. Now Wang et al. employ a bisthienothiophene conjugated linker along with a hydrophobic triphenylamine electron-donor and a hydrophilic cyanoacrylic acid electron-acceptor to construct a high molar extinction coefficient organic photosensitizer. In conjunction with a solvent-free ionic liquid electrolyte, they have fabricated a 6.5% cell exhibiting an excellent stability measured under thermal and light-soaking dual stress. |
The dye-sensitized solar cell (DSC) has been regarded as one of the most promising technologies toward cost-effective solar energy exploitation.1 In the last two decades, enormous efforts have greatly lifted the performance of DSCs, now reaching a respectable high efficiency2 up to 11.0–11.3% measured under standard air mass 1.5 global (AM 1.5G) sunlight, and a good stability3 under prolonged light and thermal dual stress. Apart from the contribution from high-quality mesoporous titania films and high-performance electrolytes, we have witnessed a pivotal role of sensitizers in this development. It is noted that so far all the record performances of DSCs were achieved using some polypyridyl ruthenium dyes.2,4,5
In view of the limited ruthenium resource, metal-free organic dyes with a high molar extinction coefficient have attracted considerable attention in the recent years owing to their low-cost and good flexibility in molecular tailoring.6 In this context, a very impressive efficiency of 9.5% has been reached with an indoline dye in conjugation with a volatile acetonitrile-based electrolyte.6j However, our preliminary tests show that it is hard to make a long-term stable device mainly due to desorption of these indoline dyes6b,j from titania nanocrystals even in conjugation with several state-of-the art stable electrolytes. Inspiringly, stable devices showing 6∼7% efficiencies have been achieved with some cyanoacrylic acid dyes.6e,g,h,i
On the basis of our systematic work on the synthesis of donor-π conjugated unit-acceptor (D–π–A) dyes,7 we have identified that dihexyloxy-substituted triphenylamide is a good donor in the construction of amphiphilic organic sensitizers, considering the feasibility of purification and the ability to suppress charge recombination. In our previous paper,7 we synthesized the C206 sensitizer (Fig. 1) with a fused thienothiophene as the π conjugated unit. Here we employ a bisthienothiophene linker to prepare a new sensitizer coded C211, with the motivation to further enhance the light-harvesting capacity and get an insight into the optoelectronic properties of these two chromophores. The synthetic details of the C211 dye are described, see ESI†.
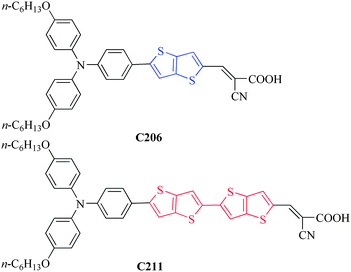 | ||
| Fig. 1 Molecular structures of the C206 and C211 photosensitizers. | ||
The electronic absorption and emission spectra of the C211 dye dissolved in chloroform are presented in Fig. 2. While the trivial ultraviolet absorption for DSCs peaks at 387 nm, the molar extinction coefficient of the low energy band at 524 nm, which mainly stems from the intramolecular charge-transfer transition, is 47 × 103 M−1 cm−1 in comparison to that of 42 × 103 M−1 cm−1 at 516 nm for C206.7 We note that along with the increase of the conjugation length, the charge-transfer transition absorption is not only bathochromic but also enhanced. The emission of the C211 sensitizer in chloroform is centered at 730 nm. The excitation transition energy (E0−0) was roughly estimated to be 2.046 eV by taking the crossing-point of the absorption and emission spectra. The C211 dye anchored on a mesoporous titania film shows a hypsochromic absorption compared to that in chloroform, suggesting that the carboxylate–titanium assembly is a weaker acceptor in contrast to the carboxylic acid.
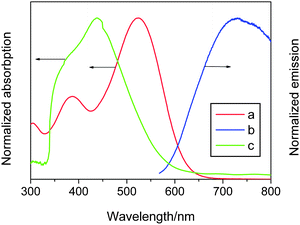 | ||
| Fig. 2 Electronic absorption (a) and emission (b) spectra of the C211 dye dissolved in chloroform. Curve (c) is the absorption spectrum of the C211 dye absorbed on a transparent titania film. The spectrum of a TiO2 reference film was subtracted for clarity in the latter. | ||
The origin of these transitions was detailed by calculating the electronic states of C211 using the time-dependent density functional theory (TDDFT) method in a Gaussian03W program suite. The absorption peak at 524 nm is mainly attributed to π → π* transitions from highest occupied molecular orbital (HOMO) to lowest unoccupied molecular orbital (LUMO) and HOMO−1 to LUMO while the absorption at 387 nm is due to electron transitions from HOMO to LUMO+1, HOMO−1 to LUMO, and HOMO−2 to LUMO. Obviously, HOMO of the C211 dye is mainly populated over the substituted triphenylamine and its adjacent thienothiophene unit while LUMO is delocalized through both cyanoacrylic acid and its adjacent thienothiophene group, with a sizeable contribution from the latter. This orientationally spatial separation of HOMO and LUMO is an ideal condition for dye-sensitized solar cells, which not only facilitates the ultrafast interfacial electron injection from the excited dye molecules to the TiO2 conduction band, but also slows down the recombination of injected electrons in TiO2 with the oxidized dye molecules due to their remoteness. In addition, the hole localized on the triarylamine unit will be spatially convenient for the electron donor to approach, facilitating fast dye regeneration.
A sensitizer must form energy offsets with the mesoporous titania and the iodide electron donor, i.e. two “type-II” heterointerfaces (titania/dye and dye/electrolyte) to overcome the exciton binding energy in an organic material for charge generation. Note that an excited dye can be an electron donor as well as an electron acceptor. A preferential charge generation at the dye/titania interface is endowed by direct chemical bonding of sensitizers; while a charge transfer at the dye/electrolyte interface is expected to be relatively slower, partially due to a physical contact. Due to the sluggish electron transfer kinetics of iodide on the normal Pt or fluorine-doped SnO2 (FTO) electrode, we coated a FTO electrode using the thermal platinization method8 to considerably reduce the oxidation overpotential and measure its HOMO accurately. In addition, ultramicroelectrode square-wave voltammetry was used to measure the HOMO and LUMO of the C211 dye in a nitrogen-filled glovebox. The downhill energy offset by the measured LUMO (−3.53 eV vs. vacuum) of the C211 dye relative to the conduction band edge (−4.00 eV1bvs. vacuum) of TiO2 provides the thermodynamic driving force for charge generation. As shown in Fig. 3, the uphill energy offset by the HOMO of C211 (−5.03 eV vs. vacuum) in contrast to that of iodide (−4.60 eV vs. vacuum), could lead to fast dye regeneration avoiding the geminate charge recombination between oxidized dye molecules and photoinjected electrons in the nanocrystalline titania film.
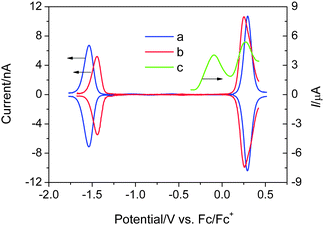 | ||
| Fig. 3 Square-wave voltammograms of a Pt ultramicroelectrode in a N,N-dimethylformide solution containing C206 (a) or C211 (b) sensitizer and 0.1 M n-tetrabutylammonium hexafluorophosphate as the supporting electrolyte. The square-wave voltammogram of 1-ethyl-3-methylimidazolium iodide (c) in 3-methoxypropionitrile with 0.1 M 1-ethyl-3-methylimidazolium bis(trifluorosulfonyl)imide supporting electrolyte was recorded with a thermally platinized FTO electrolyte. The LUMO and HOMO were estimated vs. vacuum: ELUMO/HOMO = −4.88−Fϕredox.9 | ||
We also compared the measured electronic energy levels of C206 and C211 dyes as well as the TDDFT calculation data. As depicted in Fig 4, adding one more thienothiophene unit not only drives up the HOMO level slightly but also drives down the LUMO, evidently narrowing the HOMO and LUMO gap due to a stronger influence on LUMO. It is also noted that the electrochemical energy gap (1.41 eV) of C211 is 0.3 eV larger than the estimated (1.71 eV) from the photocurrent action spectrum shown in Fig. 5A. This difference is a common feature of organic optoelectronic materials10 and may be related to the solvent effect.
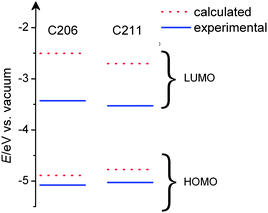 | ||
| Fig. 4 Calculated and measured energy diagram of C206 and C211 dyes. | ||
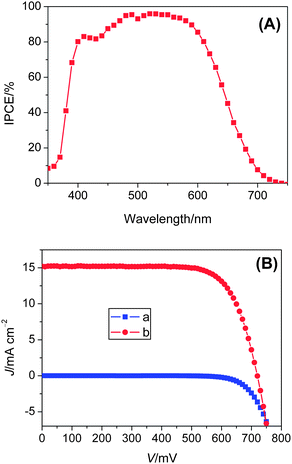 | ||
| Fig. 5 (A) Photocurrent action spectrum and (B) J–V characteristics of a DSC with the C211 sensitizer in conjugation with an acetonitrile-based electrolyte in the dark and under illumination of AM 1.5G sunlight (100 mW cm−2). Aperture area of the testing mask: 0.158 cm2. A UV-absorbing antireflection film is covered on the cell during measurements. | ||
Some preliminary photovoltaic tests were conducted to evaluate the potential of the C211 sensitizer in dye-sensitized solar cells. A screen-printed double layer film of interconnected TiO2 particles was used as a mesoporous negative electrode. A 7 µm thick film of 20 nm-sized TiO2 particles was first printed on the FTO conducting glass electrode and further coated by a 5 µm thick second layer of 400 nm-sized light scattering anatase particles. Detailed preparation procedures for TiO2nanocrystals, pastes for screen-printing, and double-layer nanostructured TiO2 films are reported in a previous paper.11 The TiO2electrode was stained by immersing in a dye solution containing 300 µM C211 and 2 mM 3α,7α-dihyroxy-5β-cholic acid co-adsorbent in chlorobenzene for 5 h. After washing with chlorobenzene (twice) and drying by air flow, the sensitized titania electrode was assembled with a platinized conducting glass counter electrode. The electrodes were separated by a 25 µm thick Surlyn hot-melt gasket and sealed by heating. The internal space was filled with a liquid electrolyte using a vacuum back-filling system. The electrolyte-injecting hole on the counter electrode glass substrate, made with a sand-blasting drill, was heat-sealed using a Surlyn sheet and a thin glass cover. Two electrolytes were used for device evaluation. Electrolyte in devices: (A), 1.0 M DMII, 50 mM LiI, 30 mM I2, 0.5 M tert-butylpyridine, and 0.1 M GNCS in the mixed solvent acetonitrile and valeronitrile (v/v, 85/15);2c (B), DMII/EMII/EMITCB/I2/NBB/GNCS (molar ratio: 12 : 12 : 16 : 1.67 : 3.33 : 0.67).3b
The photocurrent action spectrum of device (A) with the C211 sensitizer is shown in Fig. 5(A). The incident photon-to-collected electron conversion efficiency (IPCE) exceeds 80% from 400 to 610 nm, reaching the maximum of 96% at 530 nm. The maximum efficiency of absorbed photon-to-collected electron conversion efficiency (APCE) is unity over a broad spectrum range in view of the light absorption and reflecting loss of the conducting glass. As shown in Fig. 5(B), the short-circuit photocurrent density (Jsc), open-circuit photovoltage (Voc) and fill factor (FF) of the cell under AM 1.5G condition (100 mW cm−2) are 15.2 mA cm−2, 720 mV and 0.733, respectively, yielding an overall conversion efficiency (η) of 8.02%. From the overlap intergral of the IPCE curve with the standard global AM 1.5G solar emission spectrum, a short-circuit photocurrent density (Jsc) of 15.1 mA cm−2 is calculated, which is in excellent agreement with the measured photocurrent density. Therefore, there is a negligible spectral mismatch between our solar simulator and standard AM 1.5G sunlight. Note that device parameters of a reference cell made with the C206 sensitizer are 13.9 mA cm−2, 731 mV, 0.740 and 7.5%. This comparative test has clearly demonstrated the merit of employing bisthienothiophene linker to enhance light-harvesting and improve the photocurrent.
As presented in Fig. 6, a fresh cell employing the C211 dye in combination with a solvent-free ionic liquid electrolyte has Jsc of 13 mA cm−2, Voc of 664 mV, FF of 0.75 and η of 6.5% under irradiation of AM 1.5G sunlight. After one week ageing, the device efficiency is even higher, up to 6.8%. This cell shows good stability, keeping 92% of its initial efficiency after 1000 h full sunlight-soaking at 60 °C. While the photocurrent is very stable, the small attenuation of device efficiency is due mainly to a drop in open-circuit photovoltage. The latter is closely related to the augmentation of surface state of the mesoporous titania film.4 We are now trying to clarify the detailed chemical nature of these states and search for new strategies to address this common issue encountered by DSCs under prolonged thermal stress.3,4
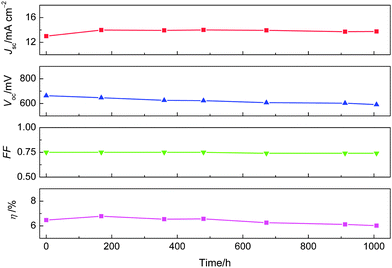 | ||
| Fig. 6 Detailed photovoltaic parameters of device (B) with a solvent-free ionic liquid electrolyte measured under irradiation of AM 1.5G sunlight (100 mW cm−2) during successive full sunlight soaking at 60 °C. | ||
In summary, we have synthesized a high molar extinction coefficient organic sensitizer showing 8% efficiency measured under AM 1.5G sunlight. In conjunction with a solvent-free ionic liquid electrolyte, we have fabricated a 6.5% cell which exhibits excellent stability measured under thermal and light-soaking dual stress. In view of this desirable spectral enhancement, we are now actively searching new concepts to boost the performance of dye-sensitized solar cells with cost-effective, metal-free organic chromophores.
This work is supported by the National Key Scientific Program – Nanoscience and Nanotechnology (No. 2007CB936700) and the “100-Talent Program” of the Chinese Academy of Sciences. Y.B. is grateful to EPFL for a visiting student fellowship. S.W., S.M.Z. and M.G. thank the Swiss National Science Foundation for financial support.
References
- (a) B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS; (b) M. Grätzel, Nature, 2001, 414, 338 CrossRef CAS; (c) N. S. Lewis, Science, 2007, 315, 798 CrossRef CAS.
- (a) M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS; (b) Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Han, Jpn. J. Appl. Phys. Part 2, 2006, 45, L638 CrossRef CAS; (c) F. Gao, Y. Wang, D. Shi, J. Zhang, M. Wang, X. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 10720 CrossRef CAS.
- (a) P. Wang, S. M. Zakeeruddin, J.-E. Moser, M. K. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402 CrossRef CAS; (b) Y. Bai, Y. Cao, J. Zhang, M. Wang, R. Li, P. Wang, S. M. Zakeeruddin and M. Grätzel, Nat. Mater., 2008, 7, 626 CrossRef CAS.
- D. Shi, N. Pootrakulchote, R. Li, J. Guo, Y. Wang, S. M. Zakeeruddin, M. Grätzel and P. Wang, J. Phys. Chem. C, 2008, 112, 17046 CrossRef CAS.
- H. J. Snaith, A. J. Moule, C. Klein, K. Meerholz, R. H. Friend and M. Grätzel, Nano Lett., 2007, 7, 3372 CrossRef CAS.
- (a) K. Hara, M. Kurashige, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, New J. Chem., 2003, 27, 783 RSC; (b) T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218 CrossRef CAS; (c) D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. Sun, Chem. Commun., 2006, 2245 RSC; (d) S. Kim, J. W. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. De Angellis, D. Di Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701 CrossRef CAS; (e) Z.-S. Wang, Y. Cui, K. Hara, Y. Dan-oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138 CrossRef CAS; (f) Z.-S. Wang, N. Koumura, Y. Cui, M. Takahashi, H. Sekiguchi, A. Mori, T. Kubo, A. Furube and K. Hara, Chem. Mater., 2008, 20, 3993 CrossRef CAS; (g) H. Qin, S. Wenger, M. Xu, F. Gao, X. Jing, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 9202 CrossRef CAS; (h) M. Wang, M. Xu, D. Shi, R. Li, F. Gao, G. Zhang, Z. Yi, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, Adv. Mater., 2008, 20 DOI:10.1002/adma.200801178; (i) Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and Kohjiro Hara, J. Phys. Chem. C, 2008, 112, 17011 CrossRef CAS; (j) S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Péchy and M. Grätzel, Chem. Commun., 2008, 5194 RSC.
- M. Xu, R. Li, N. Pootrakulchote, D. Shi, J. Guo, Z. Yi, S. M. Zakeeruddin, M. Grätzel and P. Wang, J. Phys. Chem. C, 2008, 112 DOI:10.1021/jp8082752.
- N. Papageorgiou, W. F. Maier and M. Gratzel, J. Electrochem. Soc., 1997, 144, 876 CAS.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259 CrossRef CAS.
- M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCullouch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061 RSC.
- P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2003, 107, 14336 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, calculation and additional experimental details. See DOI: 10.1039/b817990e |
| This journal is © The Royal Society of Chemistry 2009 |