Hydroxamate anchors for water-stable attachment to TiO2 nanoparticles†
William R.
McNamara
,
Robert C.
Snoeberger III
,
Gonghu
Li
,
Christiaan
Richter
,
Laura J.
Allen
,
Rebecca L.
Milot
,
Charles A.
Schmuttenmaer
*,
Robert H.
Crabtree
*,
Gary W.
Brudvig
* and
Victor S.
Batista
*
Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107. E-mail: charles.schmuttenmaer@yale.edu; robert.crabtree@yale.edu; gary.brudvig@yale.edu; victor.batista@yale.edu
First published on 7th August 2009
Broader contextAttachment of photoactive molecules to semiconductor TiO2 nanoparticles is the basis of Grätzel dye-sensitized solar cells and of a number of proposed water-splitting solar cells. Many current linkers have either mediocre water-resistance or limited ability to transmit photoinjected electrons or are unstable to oxidative conditions. This paper demonstrates robust attachment via oxidation- and water-stable hydroxamate linkers that are also able to mediate photoinjection of electrons into TiO2. Hydroxamates were selected because they have long attracted attention in a biological context—they are preferred ligands in siderophores where they bind tightly to Fe(III). |
Surface functionalization of nanoparticles is of broad interest, such as for dye attachment in dye-sensitized solar cells (DSSCs)1,2 and photocatalysis.3,4 Visible-light photoexcitation of the dye gives interfacial electron transfer (IET) into the conduction band of a semiconductor host.5 In a Grätzel cell, TiO2 is functionalized with Ru polypyridyl complexes that attach via carboxylate substituents2 that permit ultrafast IET but are unstable in aqueous conditions.6 We now report on hydroxamate anchors for robust TiO2 functionalization even in aqueous conditions.
Hydroxamate ligands bind tightly to transition metals, even in water.7,8 For example, bacterial siderophores that contain hydroxamates can dissolve Fe(III) from the oxide.7 Recent studies have reported binding of hydroxamic acids to TiO2.8 Here, we investigate their potential as robust anchors for functionalization of TiO2 thin-films commonly used in solar energy conversion and photocatalysis. We synthesize and deposit a hydroxamate-functionalized terpyridine and demonstrate visible-light sensitization of TiO2 and activation of Mn adsorbates by ultrafast IET by using spectroscopy and molecular modeling.
The synthesis (Scheme 1) builds on prior methods9 and proceeds in two steps in good yield. The methyl ester (1) reacts with O-Bn hydroxylamine (BnONH2) in the presence of LiHMDS to give the corresponding ester.10 The ester is then deprotected with H2 and Pd/C to give the product 2.
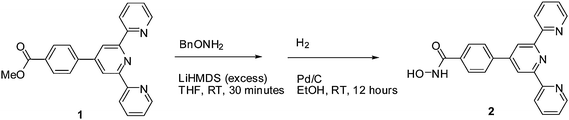 | ||
| Scheme 1 | ||
Degussa P25 TiO2 nanoparticles (NPs) were sensitized with a solution of 2 in dry EtOH using known techniques.4,11 The resulting sensitized nanoparticles were characterized using UV-visible and FTIR spectroscopy (see Fig. S1 and S2†). The spectroscopic data are consistent with 2 anchoring to TiO2via the hydroxamate. Upon binding, the disappearance of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1635 cm−1 present in the IR of unbound 2 is consistent with a −O–CRN–O− unit binding to TiO2 with terpyridine pointing away from the surface, an arrangement that is particularly suitable for the subsequent immobilization of a metal for photocatalysis. We have immobilized Mn(II) by treating the 2–TiO2 assembly with 2 mM aq. MnII(OAc)2.
O stretch at 1635 cm−1 present in the IR of unbound 2 is consistent with a −O–CRN–O− unit binding to TiO2 with terpyridine pointing away from the surface, an arrangement that is particularly suitable for the subsequent immobilization of a metal for photocatalysis. We have immobilized Mn(II) by treating the 2–TiO2 assembly with 2 mM aq. MnII(OAc)2.
Fig. 1a shows the characteristic EPR spectra at 6 K of a Mn(II) complex incorporated into 2–TiO2. In the dark, the Mn(II) signal is clearly visible (red) and, under illumination with visible light (λ ≥ 420 nm), the Mn(II) signal decreases (blue) indicating Mn(II) photooxidation to Mn(III). After the light is turned off, the Mn(II) signal fully recovers, indicating reversible photoinduced charge separation. Fig. 1 (right) shows the reversible photooxidation of Mn(II) with a 44 s half-life for e−/h+ recombination, indicating slower charge recombination than for the analogous reaction with catechol or acetylacetonate anchors.4,11 These results demonstrate visible-light oxidation of Mn(II) adsorbates through IET via the hydroxamate anchor into the TiO2 conduction band.
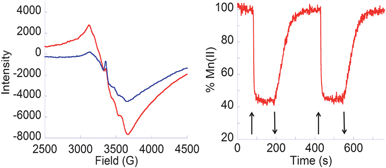 | ||
Fig. 1
Left: EPR spectra at 6 K of 2–TiO2 NPs functionalized with MnII(OAc)2 in the dark before irradiation ( ) and with visible light irradiation ( ) and with visible light irradiation ( ); Right: Time-dependent Mn(II) %age at 3106 G with light on (↑) and off (↓). ); Right: Time-dependent Mn(II) %age at 3106 G with light on (↑) and off (↓). | ||
To estimate the IET time scales and compare hydroxamate linkers to the corresponding carboxylate analogs, we simulated the IET dynamics in fully atomistic models of functionalized TiO2 nanostructures, as previously reported,4,11,12 according to mixed quantum-classical molecular dynamics. The simulations predict that both linkers lead to ultrafast, subpicosecond, IET rates and that hydroxamate anchors could replace carboxylates without altering the charge separation efficiency. Fig. 2 shows the survival probability for the electron to remain on the photoexcited adsorbate, along with a snapshot of the electronic charge distribution at 100 fs after photoexcitation: more than 50% of the electron charge is injected within the first 100 fs of dynamics.
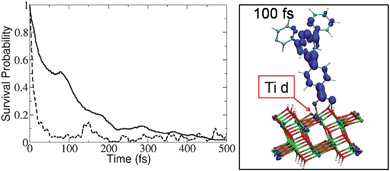 | ||
Fig. 2
Left: Time-dependent probability of the electron to remain in the adsorbate 2 (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), compared to the analogous carboxylate-linked adsorbate ( ), compared to the analogous carboxylate-linked adsorbate (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ). Right: A snapshot of the electronic charge distribution at 100 fs after photoexcitation of 2. ). Right: A snapshot of the electronic charge distribution at 100 fs after photoexcitation of 2. | ||
With time resolved terahertz spectroscopy (TRTS) we have measured the time scale and efficiency of charge injection into TiO2 NPs functionalized with 2 (Fig. 3). These measurements have subpicosecond temporal resolution and exploit the fact that mobile electrons absorb THz radiation when injected into the TiO2 conduction band.13 The THz transmission amplitude decreases when electrons are injected into the TiO2 conduction band upon adsorbate photoexcitation. Fig. 3 shows the time dependent change in THz transmittance due to ultrafast interfacial electron injection induced by 400 nm photoexcitation of TiO2 NP colloidal thin films functionalized with 2 (black) and the carboxylate-linked dye N7192 (red). While the transmission unit in Fig. 3 is arbitrary, we have used the same scaling factor for both spectra to make meaningful amplitude comparisons between 2 and N719. The rapid decrease in THz transmission indicates that IET from the adsorbate to the NP is completed on a subpicosecond time scale after photoexcitation of the system. We note that the THz absorbance signal has a fast decay component that lasts ∼10 ps for both sensitizers and then levels off. This indicates that, within 10 ps, a fraction of the injected electrons either get trapped at TiO2 defects or surface sites, or recombine with oxidized adsorbates. In addition, we have calculated the apparent injection efficiency of 2 relative to N719 with 400 nm excitation. Fig. 3 shows that 1.95 times as many electrons are injected with 2 relative to N719. At 400 nm, the absorbance of the sample functionalized with 2 is 0.67 and that for the N719 sample is 0.81 (Fig. S3†), and both films have the same thickness of 11.3 µm. The surface coverage is 1.20 and 0.99 molecules/nm2 for 2 and N719, respectively (Table S1†). Therefore, given that there is a higher coverage of 2, but a lower amount of absorbance, the fraction of molecules of 2 that have absorbed a photon is (0.99/1.20) × (0.67/0.81) = 0.68 times that of N719 (yet 1.95 times as many electrons are injected). Therefore, on a per molecule basis, 2 is 1.95/0.68, or 2.9 times more efficient than N719 for electron injection when exciting with 400 nm light. Since N719 is thought to be 100% efficient, this should not be taken to mean that we have an efficiency > 100%. The THz data only records mobile injected electrons and thus we only argue that hydroxamate is not inefficient for IET.
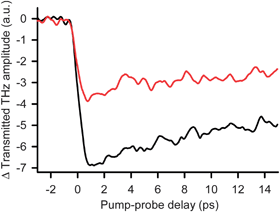 | ||
Fig. 3 400 nm pump/THz probe of electron injection in functionalized NP films of N719-TiO2 ( ) and 2-TiO2 (). ) and 2-TiO2 (). | ||
The stability of hydroxamate anchors has been compared to a carboxylate analogue via binding enthalpy calculations and UV-visible spectroscopic data. The calculations suggest that hydroxamates are ca. 33% more stable (∼10 kcal mol−1) than carboxylates on TiO2 anatase. Possible reasons are the dianionic charge, the less strained hydroxamate bite angle (∼75°) versus chelating carboxylate (∼61°) and the higher pKas for hydroxamic acids (8.88 for MeCONHOH)8aversus carboxylic acids (4.76 for MeCOOH). Experimentally, hydroxamate anchors are more stable to water. Fig. 4 (left) compares the UV-visible spectrum of 2–TiO2 to that of bare TiO2. The absorbance of 2–TiO2 at 420 nm likely corresponds to visible light excitation of an electron in the ligand HOMO to the conduction band of TiO2. Fig. 4 (right) compares the absorption at 450 nm (blue trace, 2–TiO2) and 550 nm (red trace, N719–TiO2) as a function of incubation time. Even after 24 h water exposure, there is no significant detachment of 2 (see also Fig. S3†). In contrast, the analogous experiment for N719 dye2 shows a rapid decay of the absorbance.
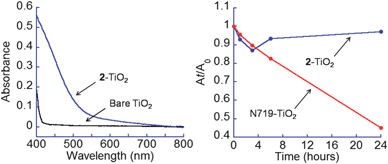 | ||
Fig. 4
Left: UV-visible spectra of 2–TiO2 ( ) and bare TiO2 (). Right: Amounts of 2 and N719 remaining attached to TiO2 NPs as a function of water incubation time from the absorbance At, at time t, relative to the A0. The wavelengths used were 450 nm for 2–TiO2 and 550 nm for N719–TiO2. ) and bare TiO2 (). Right: Amounts of 2 and N719 remaining attached to TiO2 NPs as a function of water incubation time from the absorbance At, at time t, relative to the A0. The wavelengths used were 450 nm for 2–TiO2 and 550 nm for N719–TiO2. | ||
The ease of synthesis, ability to induce ultrafast IET, and superior binding properties make hydroxamate anchors promising linkages for functionalizing TiO2 thin-films suitable for DSSCs and photocatalysis and may lead to dye-sensitized solar cells that are more resistant to humidity.
Acknowledgements
We acknowledge support from the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy (DE-FG02-07ER15909) and DOE supercomputer time from NERSC. Also, NSF ECCS-0404191 grant supported preliminary work.References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Muller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- (a) N. Serpone, Res. Chem. Intermed., 1994, 20, 953–992; (b) A. Mills and S. Leunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- S. G. Abuabara, C. W. Cady, J. B. Baxter, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig and V. S. Batista, J. Phys. Chem. C, 2007, 111, 11982–11990 CrossRef CAS.
- (a) Y. X. Weng, Y. Q. Wang, J., B. Asbury, H. N. Ghosh and T. Q. Lian, J. Phys. Chem. B, 2000, 104, 93–104 CrossRef CAS; (b) I. Martini, J. H. Hodak and G. V. Hartland, J. Phys. Chem. B, 1999, 103, 9104–9111 CrossRef CAS.
- A. D. Weisz, A. E. Regazzoni and M. A. Blesa, Solid State Ionics, 2001, 143, 125–130 CrossRef CAS.
- C. Cocozza, C. C. G. Tsao, S. F. Cheah, S. M. Kraemer, K. N. Raymond, T. M. Miano and G. Sposito, Geochim. Cosmochim. Acta, 2002, 66, 431–438 CrossRef CAS.
- (a) J. Yang, P. J. Bremer, I. L. Lamont and A. J. McQuillan, Langmuir, 2006, 22, 10109–10117 CrossRef CAS; (b) H. G. Upritchard, J. Yang, P. J. Bremer, I. L. Lamont and A. J. McQuillan, Langmuir, 2007, 23, 7189–7195 CrossRef CAS.
- (a) L. De Luca, G. Giacomelli and M. Taddei, J. Org. Chem., 2001, 66, 2534–2537 CrossRef CAS; (b) M. P. Sibi, H. Hasegawa and S. R. Ghorpade, Org. Lett., 2002, 4, 3343–3346 CrossRef CAS.
- A. Gissot, A. Volonterio and M. Zanda, J. Org. Chem., 2005, 70, 6925–6928 CrossRef CAS.
- W. R. McNamara, R. C. Snoeberger, G. Li, J. M. Schleicher, C. W. Cady, M. Poyatos, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130, 14329–14338 CrossRef CAS.
- (a) S. G. Abuabara, L. G. C. Rego and V. S. Batista, J. Am. Chem. Soc., 2005, 127, 18234–18242 CrossRef CAS; (b) L. G. C. Rego, S. G. Abuabara and V. S. Batista, J. Chem. Phys., 2005, 122, 154709 CrossRef; (c) L. G. C. Rego and V. S. Batista, J. Am. Chem. Soc., 2003, 125, 7989–7997 CrossRef CAS.
- (a) J. B. Baxter and C. A. Schmuttenmaer, J. Phys. Chem. B, 2006, 110, 25229–25239 CrossRef CAS; (b) C. A. Schmuttenmaer, Chem. Rev., 2004, 104, 1759–1779 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, experimental procedures, and modeling. See DOI: 10.1039/b910241h |
| This journal is © The Royal Society of Chemistry 2009 |