Catalytic production of biodiesel and diesel-like hydrocarbons from triglycerides
Jairton
Dupont
*a,
Paulo A. Z.
Suarez
*b,
Mario R.
Meneghetti
*c and
Simoni M. P.
Meneghetti
c
aLaboratory of Molecular Catalysis, Institute of Chemistry, UFRGS, Avenida Bento Gonçalves, 9500, Porto Alegre, 91501-970, RS, Brazil. E-mail: jairton.dupont@ufrgs.br; Fax: +55 51 33087304; Tel: +55 51 330862321
bLaboratory of Materials and Fuels, Institute of Chemistry, UnB, Brasilia, Brazil. E-mail: psuarez@unb.br; Fax: +55 61 32734149; Tel: +55 61 37994552
cGroup of Catalysis and Chemical Reactivity, Institute of Chemistry and Biotechnology, UFAL, Av. Lourival de Melo Mota, s n°, Maceio, 57072-000, AL, Brazil. E-mail: mrm@qui.ufal.br; Fax: +55 82 32141384; Tel: +55 82 32141373
First published on 6th August 2009
Abstract
Because of their high viscosity and density, crude fats and oils extracted from animal or vegetable sources (containing mainly triglycerides) cannot be directly combusted in modern diesel engines; therefore, they must be converted into biofuels. This important transformation is currently a technological bottleneck for the generation of biofuels either by thermocatalytic cracking to produce diesel-like hydrocarbons or by alcoholysis to yield mono-alcohol fatty acid esters (biodiesel). Although both transformations are relatively simple chemical reactions (thermolysis ((hydro)cracking) and (trans)esterification of triglycerides), they have several drawbacks, mainly related to catalyst efficiency and water and energy consumption. The most recent catalytic approaches and achievements, such as alcoholysis of triglycerides under multiphase conditions using classical acid or base catalysts as well as biocatalysts, are highlighted and discussed.
 Jairton Dupont Jairton Dupont | Jairton Dupont received his PhD from the Louis Pasteur University (Strasbourg, France) and after a period as a postdoc at the University of Oxford, he became a Professor of Chemistry at the Institute of Chemistry, UFRGS (Brazil). He has been invited as a Professor at ULP, Nuremberg-Erlangen and Universidad de Alcala de Henares. He is a member of the Brazilian Academy of Sciences and has received the Humboldt Research Award. His research interests are centered on ionic liquids with special emphasis on catalysis, nanomaterials and alternative energies. He has authored around 180 scientific publications and several patents and book chapters. |
 Paulo A. Z. Suarez Paulo A. Z. Suarez | Paulo A. Z. Suarez, born in 1970 in Quarai-RS, studied at UFRGS, where he obtained his chemical engineering (1993), masters (1996) and PhD (2000) degrees in the group of Professor Dupont. He began work as a visiting scientist (2001) at the Institute of Chemistry, UnB (Brasília, Brazil) and in 2002 he became a Professor of Technological Chemistry at the same institute. He has acted as a Visiting Scientist at USDA/ARS/NCAUR in Peoria, IL, USA (2006, 2007, 2008 and 2009). His research interests are centered on biofuels and biomaterials. He has authored around 57 scientific publications and 6 patents. |
 Mario R. Meneghetti Mario R. Meneghetti | Mario Roberto Meneghetti received his PhD in Organometallic Chemistry and Inorganic Chemistry from the Louis Pasteur University (Strasbourg, France), and from the Sao Paulo State University (Araraquara, Brazil) in 1999. After two postdoctoral positions at Heidelberg University (Germany), and at the Federal University of Rio Grande do Sul (Porto Alegre, Brazil) he became, in 2002, Professor of Inorganic Chemistry at the Federal University of Alagoas, Maceió, Brazil. His research interests are centered on organometallic chemistry, catalysis, and new materials. |
 Simoni M. P. Meneghetti Simoni M. P. Meneghetti | Simoni M. Plentz Meneghetti has 15 years of experience in the chemical and petrochemical industry. She obtained her PhD in the Physico-chemistry of Macromolecular Materials at the Louis Pasteur University (Strasbourg, France), in 2000. After a postdoctoral position at the Federal University of Alagoas (Maceió, Brazil), she became a Professor of Inorganic Chemistry at the same University (2006). Her research interests are focused on catalysis and oleochemistry and she is carring out several research projects concerning the development of catalysts to obtain alternative fuels derived from vegetable oils and substitute crops to biofuels production. |
Broader contextCrude fats and oils extracted from animal or vegetable sources (containing mainly triglycerides) are currently among the most important and used raw biomaterials converted into biofuels. Although this transformation can be accomplished using relatively simple chemical reactions—thermocatalytic cracking to produce diesel-like and (trans)esterification of triglycerides to yield biodiesel, they have several drawbacks mainly related to energy and water consumption. Therefore the development of more efficient catalytic processes for these transformations is one of the key factors that inhibits the definitive implementation of such fuels globally. In this article we highlight the most recent catalytic approaches used for the alcoholysis of triglycerides and the thermolysis of crude vegetable oils. |
Introduction
Renewable energy sources are of great concern to policy makers when considering economic and geopolitical factors such as high oil prices, environmental problems and supply instability.1,2 Indeed, imperative concerns related to energy, security and global climate change require large-scale substitution of petroleum-based fuels. Due to their functional similarity to petroleum-based fuel, biofuels obtained from triglycerides, such as diesel-like hydrocarbons obtained by thermocatalytic cracking or biodiesel (mono-alcohol fatty acid esters) produced by alcoholysis,3–7 have become very attractive alternatives for diesel engines. Interestingly, peanut oil was successfully used as liquid fuel in a diesel engine in 1900 during the Paris Exposition.8 However, because of its low cost and easy availability at that time, petroleum became the dominant energy source and was developed as the primary fuel for diesel engines. Nonetheless, there have been periods of supply shortages of petroleum and its derivative fuels, and as a consequence, a search for alternative energy sources has emerged. Thus, in the 1930s and 1940s, neat vegetable oils were used in diesel engines in emergency situations.9 Indeed, a mixture of fatty acid ethyl esters was produced in Belgium by ethanolysis of palm-tree oil10 and used as fuel in diesel engines. Similarly, a mixture of hydrocarbons was produced in China by a tung oil pyrolysis batch system and used as a liquid fuel.11More recently, the use of fatty acid methyl or ethyl esters (FAME or FAEE), largely known as biodiesel, has been highlighted in different countries as the main alternative for large-scale substitution of diesel fuel. However, the use of biomass as raw material for the production of fuels creates a dilemma with respect to the balance between food security and energy. If countries like Brazil and the USA incentivise the production of the so-called first generation of biofuels such as ethanol and biodiesel, European countries and international agencies are concerned about a possible food shortage crisis due to the shift of land and commodities traditionally used for the production of food towards the production of fuels.12,13 It is likely that this may occur in some parts of the word; however, in Brazil, the situation is quite different. In fact, in 2008, only 13% of land used for agriculture in Brazil was occupied by the sugar cane complex (production of sugar and ethanol).14
The production and use of biodiesel may correspond to genuine solar energy conversion, i.e., biodiesel may be considered a renewable clean energy without CO2 emissions only if the total energy invested into the cultivation of plants, the harvest and transport of the seeds and the conversion of the seeds into biodiesel is lower than the energy of the biodiesel produced (life cycle assessment (LCA) > 1). It is obvious that this carbon-neutral situation can be more easily attained by minimising energy consumption in all processes, especially in the production of plant seeds and in the chemical processes used to convert the plant seed components into biofuels. Although conversion of waste vegetable cooking oils into biofuel is an effective recycling method, the amount of biofuel thus produced can only supply a small part of diesel-engine vehicle demand. Although plant products such as soybeans are foods with high nutritional value, they are also an important supply of raw materials for biofuel production. However, plants bearing inedible products, such as Jatropha curcas, do not compete with foods in particular if they can be cultivated in semi-arid lands, and are thus expected to be the most important resources for the production of biofuels. Moreover, algae are also a potential source of biofuel raw material. Therefore, there is not a sole solution but rather many sources of biofuel raw materials that should be considered, taking into account various aspects related to the available technology, agriculture, sociology, geopolitics and economics.
Crude fats and oils extracted from animal or vegetal sources (containing mainly triglycerides) cannot be directly combusted in modern diesel engines due to their high viscosity and density. Consequently, these vegetable oils must be converted into biofuels using chemical processes that consume energy and water. This transformation is likely the most important technological bottleneck for the generation of biofuels either by thermocatalytic cracking to produce diesel-like hydrocarbons or by alcoholysis to yield mono-alcohol fatty acid esters (biodiesel). In both approaches, the glycerine fragment of triglycerides, which links three long-alkyl chains, is removed, leading to compounds containing at most one-third of the original molecular weight. Although both transformations are relatively simple chemical reactions (thermolysis ((hydro)cracking, Scheme 1) and (trans)esterification of triglycerides (Scheme 2)), they have several drawbacks, mainly related to catalyst efficiency and water and energy consumption. We will highlight and discuss the recent achievements and approaches used to overcome these drawbacks, in particular the design of catalysts mainly developed in our laboratories over the past 7 years.
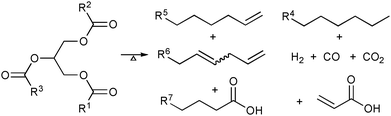 | ||
| Scheme 1 Simplified representation of several products resulting from the thermolysis of triglycerides. | ||
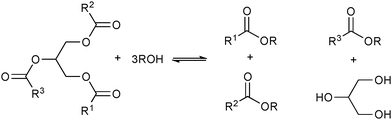 | ||
| Scheme 2 Simplified representation of the products (biodiesel and glycerine) obtained from the alcoholysis of triglycerides. | ||
Cracking and hydrocracking of triglycerides
The pyrolysis of fatty acid based materials leads to a mixture of many hydrocarbons and oxygenated compounds (Scheme 1). Indeed, the mixture produced by pyrolysis of tung oil in a batch system was used as liquid fuel in China as a hydrocarbon supply during World War II.11 In the recent decades, numerous methods for thermal15–18 and catalytic pyrolysis under inert19–26 or reductive27 atmosphere conditions have been developed. For a recent review of the development of catalysts for fatty-based materials see Suarez et al.28The characterisation of gas and liquid products obtained in those research works showed that the pyrolysis leads to not only the desirable linear and cyclic paraffins and olefins but also to undesirable oxygenated compounds, such as aldehydes and carboxylic acids. It was presumed that these oxygenated compounds are produced when the glyceridic part of the triglyceride is cracked.27 It was suggested that the reaction occurs in two steps: (i) triglycerides are decomposed to achieve carboxylic acids, acrolein and ketenes, which recombine at the reaction conditions to afford esters, carboxylic acids and hydrocarbons; (ii) the carboxylic acids are decarboxylated or decarbonylated, producing, respectively, carbon dioxide and paraffins or carbon monoxide, olefins and water. It was also pointed out in these works that the composition of the pyrolysis product can be tuned by the use of different raw materials or by the presence of heterogeneous catalysts.
It was recently observed that it is possible to isolate a fuel with physicochemical properties comparable to those specified for petroleum diesel by pyrolysis of edible soybean oil and palm-tree oil29 and also of industrial fatty wastes, such as soybean soapstock, beef tallow and poultry industry waste in the absence of catalysts.30 In all cases, a diesel-like fuel was isolated by fractional distillation of the resulting mixture of hydrocarbons and oxygenated compounds by an adequate choice of temperature intervals. It is important to highlight that the main physicochemical properties of those isolated diesel-like fuels (density, viscosity, distillation curve, carbon residue, copper corrosion test, cetane index, cold finger plugging point and heating value) were determined using ASTM standard methods and matched Brazilian specifications for diesel fuel.
The fuel resulting from the above processes possesses remaining undesirable carboxylic acid content, which was determined by GC-FID, GC-MS and acid number (ASTM D465-9). Thus, we studied the catalytic upgrading of these mixtures using soybean pyrolytic fuel. In our first approach, the vapour feed leaving the pyrolysis reactor was forced to pass through a 2 cm H-ZMS5 zeolite plug at 400 °C before its condensation.29 The heavy fraction was analysed by gas chromatography and compared with its non-deoxygenated analogue, showing that the peaks corresponding to carboxylic acids diminished or vanished completely and that new peaks emerged, corresponding to hydrocarbon products. This result clearly indicates that it is possible to deoxygenate the vegetable oil pyrolysis product in order to obtain an enriched hydrocarbon diesel-like fuel.
As a second approach to improve the deoxygenation of these biofuels, we studied the pyrolysis of soybean oil in the presence of the solids (SnO)2(Al2O3)8, (SnO)(ZnO)(Al2O3)8 and (ZnO)2(Al2O3)831 and, after adequate distillation, analysed the pyrolytic products by GC-FID, GC-MS and the acid number in order to determine the catalyst with the highest product deoxygenating efficiency. It was observed by the acid number, FT-IR, GC-FID and GC-MS that the nature of the solid influences the quantity of acid products, which can be related to the activity of the catalyst in deoxygenating the pyrolysis products. These results suggest the following order for the activity of the catalysts towards the decomposition of the carbonyl compounds during pyrolysis: (SnO)(ZnO)(Al2O3)8 > (SnO)2(Al2O3)8 > (ZnO)2(Al2O3)8. It is also important to highlight that only small differences in the distillation temperature fraction distribution were observed, especially for the diesel-like fraction, which likely means that these catalysts are not active in the carbon chain cracking process. More studies are currently under way in our laboratories to improve the catalytic activity of doped alumina in the deoxygenation of the pyrolysis products of triglycerides and to determine the possibility of recovering these catalysts.
It is important to highlight that the use of pyrolysis or catalytic cracking is still controversial due to concerns about its industrial coasts. Indeed, two different arguments are usually used: (i) high energy consumption in order to process the reaction at high temperatures; and (ii) low reaction yield in diesel-like product (around 70%) when compared to mono-esthers production (up to 95%). It is difficult to answer these two arguments as far as this technology is still not being used in industrial scale and few research efforts have been done when compared with other biofuels technologies. Indeed, there are only few examples of large-scale use of cracking of vegetable oils have been reported in the literature. One example was the Chinese experience with tung oil using regular refineries during the 1940's and the other one was the H-BIO patented by PETROBRAS.32 In the PETROBRAS experience (Fig. 1), soybean oil (up to 20%) was mixed with a pool of diesel before hydrodesulfurisation process (HDT), being produced water, carbon mono and dioxides, and long chains saturated hydrocarbons, methane and propane.
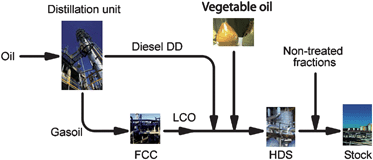 | ||
| Fig. 1 Simplified scheme of the H-BIO process that may employs crude vegetable oils. | ||
It is interesting to note that these two experiences used the same industrial plants used to process petroleum, which means that there is no need to build new industrial facilities to produce biofuels from cracking fatty materials. However, one should take in account that the temperature for cracking fatty materials is lower than those usually needed to crack petroleum, which allow us to think that it energy balance in large scale is probably better. On the other hand, it should also be noted that the purification of the final product is simpler than first generation biodiesel, which can be done just leading the products from the cracking reactor into a plate tower. We have recently set up a pilot plant and found out that its energy balance, even in small scale, is 3 to 1, close to those verified in petroleum refineries. Moreover, it is possible to use low cost and low quality raw materials, such as high acid fats and oils, industrial or domestic fatty acids, which are almost impossible to transform using traditional technologies, to produce high-quality biofuels.33
Alcoholysis of triglycerides using Brønsted acid and base catalysts
The alcoholysis (in particular, methanolysis) of vegetable oils catalysed by metal hydroxides (or alkoxides) and acids is the most commonly used method for the generation of biodiesel (Scheme 2).34 However, technological problems such as corrosion, emulsification and separation of glycerine and water and energy consumption are usually associated with these acid/base methodologies.6,35 Sulfuric, phosphoric or sulfonic acids are the most used acid catalysts. These acidic processes requires temperatures >100 °C for at least 3 h and molar ratio of 30 between the alcohol and the oil.9,36–38The base catalysed reactions are usually 4000 times faster then the transesterifications reactions promoted by the same quantity of acid.39 The experimental protocols in these base catalytic processes are: 6 : 1 molar ratio of methanol to oil, reaction temperatures between 25 and 120 °C for 0.5 to 6 h. In these cases the starting oil should have low levels of acidity (less then 0.5% of free acids) and moisture should be avoided.3,40
Although the base-catalysed reaction is faster and requires lower operating temperatures than the acid-catalysed reaction, acid catalysis is more efficient when the amount of free fatty acids in the oil exceeds 1%. Indeed, economically, the acid-catalyzed procedure, being a one-step process, is more economical than the alkali-catalyzed process that requires an extra step to convert free fatty acids to methyl esters in order to avoid soap formation.34,41
However, these catalytic processes are restricted to methanol and cannot be directly transposed to use with the lower-cost ethanol and fusel oil, which is a by-product of the distillation of ethanol from fermentation of molasses and which contains mainly C3–C5 alcohols.
Separable and reusable heterogeneous catalysts have been studied to decrease the environmental impact of the production of biofuels. Indeed, active and stable heterogeneous catalysts for esterification, transesterification and hydrolysis may provide promising processes for environmentally benign production of biofuels.42 In particular, amorphous carbon bearing SO3H moieties43,44 or nanotubes with double acid sites (i.e, Brønsted acid and Lewis acid sites)45 functions as a highly active and stable solid catalyst for various acid-catalysed reactions such as direct conversion of crude vegetable oils into biodiesel and glycerol. These effective, inexpensive, and reusable polycyclic aromatic carbon acid catalysts can be prepared by partial carbonization and sulfonation of glycerol46 or sugar43 in presence of sulfuric acid. In the “supported-sulfuric acid” transesterification process, the crude oils are converted into a mixture of higher fatty acids, glycerol and water by the hydrolysis of triglycerides, and the higher fatty acids are then transformed into biodiesel through esterification. This method does not require the costly and energy-inefficient dehydration processes nor does it generate waste usually associated with the classical sulfuric acid catalysed processes. However, separation of the glycerol, the use of large amounts of alcohol (mainly restricted to methanol) and the relatively high temperature (403 K) required are major drawbacks of these solid acid catalytic processes. A more efficient transesterification (alcoholysis) catalytic system was obtained by the immobilisation of the classical basic or acid catalysts in ionic liquids.47–50 For example, the combination of the ionic liquid 1-n-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (BMI·NTf2, Fig. 1)), alcohols and inorganic bases such as M2CO3 (M = K or Cs) results in the production of biodiesel from soybean oil in high yields (>98%) and purity. Classical H2SO4 immobilised in the same ionic liquid efficiently promotes the transesterification reaction of soybean oil and various primary and secondary alcohols. In this multiphase process, the acid is almost completely retained in the ionic liquid phase, while the biodiesel forms a separate phase. The recovered ionic liquid containing the acid could be reused at least six times without any significant decrease in biodiesel yield or selectivity. In both catalytic processes (acid and base), the reactions proceed as a typical multiphase system, in which the formed biodiesel accumulates in the upper phase, and the glycerol by-product is selectively captured by the alcohol-ionic liquid-acid/base phase. This glycerol extraction–during the (trans)esterification–by the ionic liquid/alcohol mixture shifts the equilibrium to the biodiesel product (Scheme 2), thus increasing the biodiesel yield. It should be noted that the solubility of glycerol in pure ionic liquid is very low (>1 wt%); however, it can reach 15 wt% in a mixture of the ionic liquid with methanol (9 mol%). The ionic liquid/alcohol that still contains the glycerol should be washed with water for 3–4 cycles to separate the glycerol, which is then usually isolated with high purity (>98%). It should be noted that the glycerol isolated from classical acid or basic catalysis requires various purification steps to achieve desirable purity. The use of acids immobilised in ionic liquids has other important technological advantages over classical procedures, such as reduction in engine corrosion, since the acid is retained almost exclusively in the ionic phase, and catalyst recycling.
Alternatively, ultrasound-assisted transesterification reaction of vegetable oils with methanol in the presence of a potassium hydroxide catalyst can be used to improve the classical base and acid processes.51–54 In some cases, the ultrasound-based processes save energy and materials and reduce reaction time and soap formation.
Alcoholysis of triglycerides using lewis acid catalysts
To study the catalytic performances of Lewis acid metal compounds for soybean methanolysis in homogeneous conditions, we prepared complexes using the ligand 3-hydroxy-2-methyl-4-pyrate (maltolate) and the cations Sn(II)+2, Zn(II)+2, Pb(II)+2 and Hg(II)+2.55 Initially, the different complexes were tested in the methanolysis of soybean oil using mild conditions, observing that the activity order obtained (Sn+2 ≫ Zn+2 > Pb+2 ≅ Hg+2) was in accordance to the Lewis acidity order of the metals. Next, different vegetable oils, such as andiroba (Carapa guianensis), babassu (Orbignia sp.), palm tree (Elaeis sp.), piqui (Caryocar sp.) and soybean (Glycine max), the fatty acids of which have different alkyd-chain lengths and numbers of double bonds, were methanolysed using the catalytic precursor [Sn(maltolate)2] at identical reaction conditions.56 It was observed that the reaction activities are strongly influenced by the nature of the vegetable oil, leading to the inference that the methanolysis reaction is favoured by the presence of unsaturations in the carbonic chain and by its length. Alcoholysis of soybean oil was also studied using different alkyl-chain alcohols such as methanol, ethanol, propanol, iso-propanol, n-butanol, tert-butanol and cyclohexanol under similar reaction conditions. It was verified that the catalytic activities are strongly dependent on the nature of the alcohol. When the alcohol alkyl chain is linear, the reaction activities decrease with increasing chain length, and when branched ones are used, the activity decreases drastically. These results strongly suggest that a steric effect is controlling the catalytic activity.Another class of compounds used as Lewis acid catalysts for transesterifications are based on organometallic Sn(IV) compounds (Fig. 2). Indeed, butylstannoic acid (BTA), di-n-butyl-oxo-stannane (DBTO), dibutyltin diacetate (DBTDA) and dibutyltin dilaurate were tested as catalysts in the methanolysis of soybean oil at different temperatures and reaction times.57
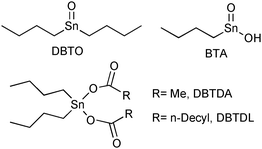 | ||
| Fig. 2 Chemical structures of organometallic Sn(IV) complexes employed as Lewis acid catalyst in the methanolysis of soybean oil. | ||
These organometallic compounds showed moderate activity for the production of fatty acid methyl esters (FAME) under mild reaction conditions (atmospheric pressure, reaction temperature around 80 °C and methanol : oil : catalyst molar ratio of 400 : 100 : 1). At those conditions, it was observed that the two most active catalysts, DBTDA and DBTDL, lead to reaction yields up to 43% and 44% after 10 h, respectively. It is worth remarking that both complexes are liquid at room temperature and soluble in methanol and in soybean oil. Conversely, the other two compounds are solid and quite insoluble in the reaction mixture. Of course, the discrepancy in solubilities of the four tested complexes can explain such a difference of catalytic performance of the catalysts at the applied reaction conditions.
However, it was observed that just replacing the former reactor by a closed steel one, a significantly improvement of reaction yields were observed. Indeed, a close reactor allows reaching reaction temperatures above of the boiling point of methanol, and also appropriate phase equilibrium is established inside the reactor. For example, after 1 h, in the presence of DBTDL, and with the same methanol : oil : catalyst molar ratio (400 : 100 : 1), up to 95% reaction yield was achieved at 160 °C, which may be related to the enhancement of the reactivity of the involved species and of the solubility of the catalyst in the reaction medium.58
Although tin complexes, especially [Sn(maltolate)2], exhibit a very good activity for vegetable oil alcoholysis, their technological potential in homogeneous conditions is very poor. Indeed, as long as these complexes remain dissolved in the reaction medium, it is difficult to recover and reuse them. For this reason, immobilisation of the tin complexes was attempted in order to obtain similar catalytic activity allied with the advantages of a heterogeneous system.59,60 To achieve this goal, two different strategies to anchor [Sn(maltolate)2] were followed: (i) in an organic solid phase; and (ii) in ionic liquids. One attempt to obtain a recyclable catalytic system was to prepare a DOWEX® acid resin containing the tin complex coordinated in the acid sites. However, when this solid was tested as a catalyst for soybean methanolysis, using the same amount of tin and similar conditions as used in the homogeneous reaction, only a 0.5% reaction yield was observed. It is worth mentioning that this reaction yield was even lower than the value observed when pure resin was used (1%). This reduction in the reaction yield can be explained by assuming that the interaction of the resin with the tin complex probably deactivated the acid sites of both the resin and the complex.59 The second attempt was a two-phase system obtained by preparing a solution of the tin complex in the ionic liquids611-n-butyl-3-methylimidazolium hexafluorophosphate (BMI·PF6, Fig. 3)59 and 1-butyl-3-methylimidazolium tetrachloroindate (BMI·InCl4, Fig. 2).60
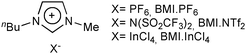 | ||
| Fig. 3 Example of imidazolium ionic liquids used for the immobilisation of catalysts for the alcoholysis of triglycerides. | ||
The reactions yields in these two-phase systems were similar to those obtained in homogeneous conditions under comparable reaction conditions. However, after recovering and reusing the ionic liquid phases, the reactions' yields drastically decreased with each new substrate charge, dropping to almost zero after the fourth one. Electrospray ionisation mass spectrometry (ESI-MS) analysis of the BMI·InCl4 ionic liquid phase and of the organic phase before and after catalysis indicate that the tin catalyst remains in the IL phase and that no leaching of the catalyst occurs.60 However, a clear decomposition of the complex was observed, which is probably the cause of the decrease in the catalytic activity during the recycle experiments.
Tin oxide was also tested as a heterogeneous catalyst for the methanolysis of soybean oil.59 Conversions of up to 56% and 94.7% were obtained after one and five hours, respectively. At the end of the reaction, it was possible to recover the catalyst by simple filtration of the mixture and by reusing the recovered solid for an additional three times under the same reaction conditions with the catalytic activity conserved.
Mixed metal-oxides of the type (Al2O3)X(SnO)Y(ZnO)Z are also active for soybean oil alcoholysis with different alkyl-chain alcohols, including branched ones.62 The best result was achieved using methanol, with conversion yields up to 80% after 4 h. As observed for Sn(maltolate)2 at homogenous conditions, the catalytic activities are strongly dependent on the nature of the alcohol. For alcohols with a linear chain, the reaction activities decrease with increasing chain length and decrease drastically when branched alcohols are used. These results strongly suggest that steric effects control these catalytic activities and that a similar mechanism to that with Sn(maltolate)2 likely takes place. The pure tin oxide solids were also recovered and reused under the same conditions, reaching similar yields after recycling the catalysts four times.
In contrast, promising surface acid catalysts for biodiesel production via transesterification reaction are ones based on sulfated metal oxides. In fact, these materials have been studied for about 20 years as potential catalysts for different types of reactions.63–65 However, just recently, sulfated ZrO2 (ZrO2/SO4) has been used in transesterification of vegetable oils, as well as in esterification of free fatty acids.66–69 Based on these results, a superacid system was developed based on an analogous structure, i.e. sulfated titania (TiO2/H2SO4). This material was prepared with titania/sulfuric acid molar ratios of 5%, and 10% (TS-5, and TS-10, respectively) and tested as catalysts in the methanolysis of soybean and castor oils. The transesterifications were carried out in a closed steel reactor at 120 °C for 1 h, with a methanol/oil/catalyst molar ratio of 120/20/1. At these conditions, the pressure of the reactor increased until reaching 5.0 bar. Unfortunately, for all catalysts, only moderate catalytic activity were observed, reaching reactions yields up to 30% to 40% when soybean oil was used, and 20% to 25% for the analogous reactions using castor oil.
Alcoholysis of triglycerides using biocatalysts
The bio(trans)esterification reaction is one promising route to environmentally benign production of biodiesel. In particular, enzymes—usually lipases isolated from microorganisms—exhibit high catalytic activity for the transesterification of triglycerides under moderate reaction conditions, resulting in high-purity biodiesel.39,70,71However, enzyme inactivation to methanol denaturation is usually observed in these processes. To solve this problem it is investigate various protocols such as by screening new resistant catalysts, including the exploration of new immobilization methods, or by optimization of the production process, namely by the application of stepwise addition of methanol or methanol prolonged released.72,73
Moreover, the glycerol formed during the transesterification of triglycerides blocks the active sites of lipase, decreasing its activity and inhibiting the reuse of the biocatalyst. One possible solution is to capture the formed glycerol using, for example, an acetyl acceptor.74 However, the main hurdle to the commercialisation of this system is the cost of lipase production, since the recovery and reuse of the enzyme is complicated even for an immobilised biocatalyst such as lipase immobilised on a solid material75 such as porous kaolinite, which functions as a reusable heterogeneous catalyst.76,77 It was found that the ionic liquid might function as both a support for the biocatalyst and as a glycerol acceptor (Fig. 4). Indeed, pseudomonas cepacia lipase supported in the 1-n-butyl-3-methylimidazoliumbis(trifluoromethyl sulfonyl) imide (BMI)·NTf2 ionic liquid catalyses the transesterification of soybean oil at room temperature in the presence of water and without the use of organic solvents.78 The ionic liquid supported lipase biocatalytic system79,80 is also compatible with various alcohols (including isoamyl alcohol). The biodiesel is separated by simple decantation, and the recovered ionic liquid/enzyme catalytic system can be reused at least four times without loss of catalytic activity or selectivity.
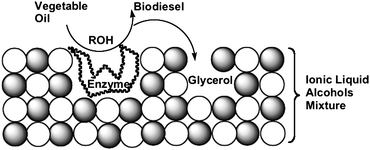 | ||
| Fig. 4 Simplified representation of the dual function (enzyme support and glycerol acceptor) of the ionic liquid in the enzymatic alcoholysis of triglycerides (adapted from ref. 58). | ||
The catalytic activities of these ionic liquid supported lipases are superior to those reported earlier for enzymatic processes in water or on other supports.70,81 This is probably a direct result of the extraction of the glycerol82—formed during the transesterification—by the ionic liquid/alcohol mixture. Interestingly, addition of water up to 5 vol% is necessary to improve the reaction rate and to increase the oil hydrolysis rate, yielding a fatty acid that is converted into its respective ester faster than by the transesterification pathway (Scheme 3).
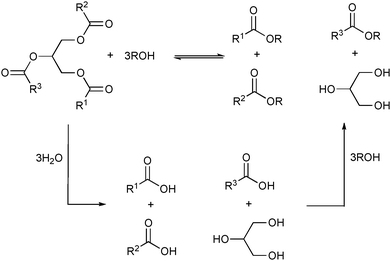 | ||
| Scheme 3 Simplified representation of several products obtained from the alcoholysis and hydrolysis followed by esterification of triglycerides. | ||
Catalytic esterification of fatty acids
The esterification of fatty acids is also an important transformation for the generation of biodiesel from acids from hydrolysis of raw biomaterials. This reaction can be autocatalytic, but without the presence of suitable catalysts the reaction yields are low. One strategy to obtain new catalytic systems for that reaction is based on the development of compounds exhibiting Lewis acid character. For this purpose, a series of Ti(IV) and Zr(IV) complexes, showing general formula M(n-butoxide)4−x(maltolate)x, were prepared (Fig. 5). The catalytic potentials of those complexes for esterification of fatty acids in the presence of methanol were evaluated.83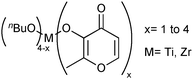 | ||
| Fig. 5 General chemical structure of M(n-butoxide)4−x(maltolate)x used as catalysts in esterification of fatty acids in the presence of methanol. | ||
It was verified that zirconium catalysts are more active than the analogous titanium ones. For example, we observed conversions up to 73.7% at 140 °C for Ti(n-butoxide)3(maltolate) and 91.3% for [Zr(n-butoxide)3(maltolate)] after 2 h. It is worth mentioning that we have also tested these complexes as catalysts for transesterification of soybean oil in the presence of methanol, and their productivities of FAME were very poor. It is highly probable that in this case, the complexes are activated only in presence of relatively acidic substrates such as fatty acids. However, most of the available fatty acid esterification catalytic systems are limited to methanol, and catalyst recovery remains a major drawback.
Conclusions
There is no doubt that both thermocracking and alcoholysis of triglycerides are important and complementary alternatives for the generation of biofuels. Although new catalytic systems have been developed in recent years, almost all of them are restricted to the methanolysis of triglycerides under “homogeneous” conditions, rendering catalyst recovery difficult. Moreover, most of these catalytic systems are energy- and water-demanding processes. Therefore, various challenges associated with these very simple transformations remain, such as for the transesterification reaction. In particular, for countries where the sugar-cane complex is economically important, the catalytic processes cannot be restricted to methanol and should be directly transposed to use the lower-cost ethanol and fusel oil. It is apparent that industrial processes will result from improvement of the catalyst efficiency intimately associated with the reaction engineering. The key factor for the solution is certainly related to the immobilisation of the catalyst (whatever its nature: acid, base or biological) that will rend the processes recyclable with the minimum use of solvents (including water) and energy. It is also evident that solid supports and ionic liquids are among the most promising potential solutions for transesterification catalyst immobilisation protocols.Acknowledgements
Thanks are due to the following Brazilian agencies for partial financial support: MCT, FINEP, CNPq, CAPES and FAPERGS.References
- D. Tilman, J. Hill and C. Lehman, Science, 2006, 314, 1598–1600 CrossRef CAS.
- M. Frondel and J. Peters, Energy Policy, 2007, 35, 1675–1684 CrossRef.
- J. M. Marchetti, V. U. Miguel and A. F. Errazu, Renewable Sustainable Energy Rev., 2007, 11, 1300–1311 CrossRef CAS.
- P. T. Vasudevan and M. Briggs, J. Ind. Microbiol. Biotechnol., 2008, 35, 421–430 CrossRef CAS.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 RSC.
- A. C. Pinto, L. L. N. Guarieiro, M. J. C. Rezende, N. M. Ribeiro, E. A. Torres, W. A. Lopes, P. A. D. Pereira and J. B. de Andrade, J. Braz. Chem. Soc., 2005, 16, 1313–1330 CAS.
- A. Sivasamy, K. Y. Cheah, P. Fornasiero, F. Kemausuor, S. Zinoviev and S. Miertus, ChemSusChem, 2009, 2, 278–300 CrossRef CAS.
- E. G. Shay, Biomass Bioenergy, 1993, 4, 227–242 CrossRef CAS.
- F. R. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- C. G. Chavanne, Belg. Pat., 422,877, Aug. 31, 1937; Chemical Abstracts 32:4313 (1938).
- C. C. Chang and S. W. Wan, Ind. Eng. Chem., 1947, 39, 1543–1548 CrossRef CAS.
- P. A. Z. Suarez, André L. F. Santos, J. P. Rodrigues and M. B. Alves, Quim. Nova, 2009, 32, 768–775 CAS.
- P. A. Z. Suarez and S. M. P. Meneghetti, Quim. Nova, 2007, 30, 2068–2071 CAS.
- http://www.ibge.gov.br/home/estatistica/indicadores/agropecuaria/lspa/lspa_200812_1.shtm, January 2009.
- J. W. Alencar, P. B. Alves and A. A. Craveiro, J. Agric. Food Chem., 1983, 31, 1268–1270 CrossRef CAS.
- I. C. P. Fortes and P. J. Baugh, J. Braz. Chem. Soc., 1999, 10 Search PubMed.
- I. C. P. Fortes and P. J. Baugh, J. Anal. Appl. Pyrolysis, 2004, 72, 103–111 CrossRef CAS.
- A. W. Schwab, G. J. Dykstra, E. Selke, S. C. Sorenson and E. H. Pryde, J. Am. Oil Chem. Soc., 1988, 65, 1781–1786 CrossRef CAS.
- F. R. Santos, J. C. N. Ferreira and S. R. R. da Costa, Quim. Nova, 1998, 21, 560–563 CAS.
- Y. S. Prasad, N. N. Bakhshi, J. F. Mathews and R. L. Eager, Can. J. Chem. Eng., 1986, 64, 278–284 CrossRef CAS.
- R. O. Idem, S. P. R. Katikaneni and N. N. Bakhshi, Energy Fuels, 1996, 10, 1150–1162 CrossRef CAS.
- R. O. Idem, S. P. R. Katikaneni and N. N. Bakhshi, Fuel Process. Technol., 1997, 51, 101–125 CrossRef CAS.
- S. P. R. Katikaneni, J. D. Adjaye, R. O. Idem and N. N. Bakhshi, J. Am. Oil Chem. Soc., 1998, 75, 381–391 CrossRef CAS.
- R. K. Sharma and N. N. Bakhshi, Can. J. Chem. Eng., 1991, 69, 1071–1081 CAS.
- L. Dandik, H. A. Aksoy and A. Erdem-Senatalar, Energy Fuels, 1998, 12, 1148–1152 CrossRef CAS.
- S. P. R. Katikaneni, J. D. Adjaye and N. N. Bakhshi, Energy Fuels, 1995, 9, 599–609 CrossRef CAS.
- J. Gusmão, D. Brodzki, G. Djega-Mariadassou and R. Frety, Catal. Today, 1989, 5, 533–544 CrossRef CAS.
- P. A. Z. Suarez, S. M. P. Meneghetti, M. R. Meneghetti and C. R. Wolf, Quim. Nova, 2007, 30, 667–676 CAS.
- D. G. Lima, V. C. D. Soares, E. B. Ribeiro, D. A. Carvalho, E. C. V. Cardoso, F. C. Rassi, K. C. Mundim, J. C. Rubim and P. A. Z. Suarez, J. Anal. Appl. Pyrolysis, 2004, 71, 987–996 CrossRef CAS.
- A. L. F. Santos, D. U. Martins, O. K. Iha, R. A. M. Ribeiro, R. L. Quirino and P. A. Z. Suarez, Agro-Industrial Residues as Low-Price Feedstock for Diesel-Like Fuel Production by Thermal Cracking, 2009, submitted for publication Search PubMed.
- R. L. Quirino, A. P. Tavares, J. C. Rubim and P. A. Z. Suarez, J. Am. Oil Chem. Soc., 2009, 86, 167–172 CrossRef CAS.
- J. R. Gomes, Braz. Pat., BR 0500591, 2005 to PETROBRAS.
- A. L. F. Santos, D. U. Martins, O. K. Iha, R. A. M. Ribeiro, R. L. Quirino and P. A. Z. Suarez, submitted for publication.
- Y. Zhang, M. A. Dube, D. D. McLean and M. Kates, Bioresour. Technol., 2003, 90, 229–240 CrossRef CAS.
- U. Schuchardt, R. Sercheli and R. M. Vargas, J. Braz. Chem. Soc., 1998, 9 Search PubMed.
- E. Lotero, Y. J. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- L. C. Meher, D. V. Sagar and S. N. Naik, Renewable Sustainable Energy Rev., 2006, 10, 248–268 CrossRef CAS.
- S. Zheng, M. Kates, M. A. Dube and D. D. McLean, Biomass Bioenergy, 2006, 30, 267–272 CrossRef CAS.
- H. Fukuda, A. Kondo and H. Noda, J. Biosci. Bioeng., 2001, 92, 405–416 CrossRef CAS.
- H. Sanli and M. Canakci, Energy Fuels, 2008, 22, 2713–2719 CrossRef CAS.
- Y. Zhang, M. A. Dube, D. D. McLean and M. Kates, Bioresour. Technol., 2003, 89, 1–16 CrossRef CAS.
- M. Di Serio, R. Tesser, L. Pengmei and E. Santacesaria, Energy Fuels, 2008, 22, 207–217 CrossRef CAS.
- M. Toda, A. Takagaki, M. Okamura, J. N. Kondo, S. Hayashi, K. Domen and M. Hara, Nature, 2005, 438, 178–178 CrossRef CAS.
- M. Hara, ChemSusChem, 2009, 2, 129–135 CrossRef CAS.
- J. Li, X. H. Wang, W. M. Zhu and F. H. Cao, ChemSusChem, 2009, 2, 177–183 CrossRef CAS.
- B. L. A. P. Devi, K. N. Gangadhar, P. S. S. Prasad, B. Jagannadh and R. B. N. Prasad, ChemSusChem, 2009, 3, 617–620 CrossRef.
- A. A. M. Lapis, L. F. deOliveira, B. A. D. Neto and J. Dupont, ChemSusChem, 2008, 1, 759–762 CrossRef CAS.
- X. Q. Chen, C. J. Liu, J. Wang and Y. P. Li, Chin. J. Org. Chem., 2009, 29, 128–134 CAS.
- Z. F. Fei, D. B. Zhao, T. J. Geldbach, R. Scopelliti and P. J. Dyson, Chem.–Eur. J., 2004, 10, 4886–4893 CrossRef CAS.
- A. A. M. Lapis, B. A. D. Neto, J. D. Scholten, F. M. Nachtigall, M. N. Eberlin and J. Dupont, Tetrahedron Lett., 2006, 47, 6775–6779 CrossRef CAS.
- F. F. P. Santos, S. Rodrigues and F. A. N. Fernandes, Fuel Process. Technol., 2009, 90, 312–316 CrossRef CAS.
- C. Stavarache, M. Vinatoru and Y. Maeda, Ultrason. Sonochem., 2007, 14, 380–386 CrossRef CAS.
- C. Stavarache, M. Vinatoru, Y. Maeda and H. Bandow, Ultrason. Sonochem., 2007, 14, 413–417 CrossRef CAS.
- C. Stavarache, M. Vinatoru, R. Nishimura and Y. Maeda, Ultrason. Sonochem., 2005, 12, 367–372 CrossRef CAS.
- F. R. Abreu, D. G. Lima, E. H. Hamu, S. Einloft, J. C. Rubim and P. A. Z. Suarez, J. Am. Oil Chem. Soc., 2003, 80, 601–604 CrossRef CAS.
- F. R. Abreu, D. G. Lima, E. H. Hamu, C. Wolf and P. A. Z. Suarez, J. Mol. Catal. A: Chem., 2004, 209, 29–33 CrossRef CAS.
- D. A. C. Ferreira, M. R. Meneghetti, S. M. P. Meneghetti and C. R. Wolf, Appl. Catal., A, 2007, 317, 58–61 CrossRef CAS.
- D. R. Mendonça, J. P. V. Silva, R. M. Almeida, C. R. Wolf, M. R. Meneghetti and S. M. P. Meneghetti, Appl. Catal., A, 2009, 365, 105–109 CrossRef.
- F. R. Abreu, M. B. Alves, C. C. S. Macedo, L. F. Zara and P. A. Z. Suarez, J. Mol. Catal. A: Chem., 2005, 227, 263 CrossRef CAS.
- B. A. D. Neto, M. B. Alves, A. A. M. Lapis, F. M. Nachtigall, M. N. Eberlin, J. Dupont and P. A. Z. Suarez, J. Catal., 2007, 249, 154–161 CrossRef.
- P. Wasserscheid, A. Metlen and N. Brausch, (Novartis A.-G., Switz.; Novartis Pharma G.m.b.H.), Application: WO, 2005, 15 pp Search PubMed.
- C. C. S. Macedo, F. R. Abreu, A. P. Tavares, M. B. Alves, L. F. Zara, J. C. Rubim and P. A. Z. Suarez, J. Braz. Chem. Soc., 2006, 17 Search PubMed.
- M. Hino, S. Kobayashi and K. Arata, J. Am. Chem. Soc., 1979, 101, 6439–6441 CrossRef CAS.
- M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1979, 1148–1149 RSC.
- G. A. Olah, G. K. S. Prakash and J. Sommer, Superacids, Wiley, New York, 2005 Search PubMed.
- S. Furuta, H. Matsuhashi and K. Arata, Biomass Bioenergy, 2006, 30, 870–873 CrossRef CAS.
- S. Furuta, H. Matsuhashi and K. Arata, Catal. Commun., 2004, 5, 721–723 CrossRef CAS.
- J. Jitputti, B. Kitiyanan, P. Rangsunvigit, K. Bunyakiat, L. Attanatho and P. Jenvanitpanjakul, Chem. Eng. J., 2006, 116, 61–66 CrossRef CAS.
- D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, J. Catal., 2007, 247, 43–50 CrossRef CAS.
- Y. Shimada, Y. Watanabe, A. Sugihara and Y. Tominaga, J. Mol. Catal. B: Enzym., 2002, 17, 133–142 CrossRef CAS.
- S. Al-Zuhair, Biotechnol. Prog., 2005, 21, 1442–1448 CrossRef CAS.
- M. M. Soumanou and U. T. Bornscheuer, Enzyme Microb. Technol., 2003, 33, 97–103 CrossRef.
- Y. Luo, G. Wang, Y. S. Ma and D. Z. Wei, J. Chem. Technol. Biotechnol., 2006, 81, 1846–1848 CrossRef CAS.
- W. Du, Y. Y. Xu, D. H. Liu and J. Zeng, J. Mol. Catal. B: Enzym., 2004, 30, 125–129 CrossRef CAS.
- W. Du, Y. Y. Xu, D. H. Liu and Z. B. Li, J. Mol. Catal. B: Enzym., 2005, 37, 68–71 CrossRef CAS.
- M. Iso, B. X. Chen, M. Eguchi, T. Kudo and S. Shrestha, J. Mol. Catal. B: Enzym., 2001, 16, 53–58 CrossRef CAS.
- P. Lozano, E. García-Verdugo, R. Piamtongkam, N. Karbass, T. De Diego, M. I. Burguete, S. V. Luis and J. L. Iborra, Adv. Synth. Catal., 2007, 349, 1077–1084 CrossRef CAS.
- M. Gamba, A. A. M. Lapis and J. Dupont, Adv. Synth. Catal., 2008, 350, 160–164 CrossRef CAS.
- Z. Guo, B. Q. Chen, R. L. Murillo, T. W. Tan and X. B. Xu, Org. Biomol. Chem., 2006, 4, 2772–2776 RSC.
- W. Du, L. Wang and D. H. Liu, Green Chem., 2007, 9, 173–176 RSC.
- Y. Watanabe, Y. Shimada, A. Sugihara and Y. Tominaga, J. Mol. Catal. B: Enzym., 2002, 17, 151–155 CrossRef CAS.
- A. P. Abbott, M. P. M. Cullis, M. J. J. Gibson, R. C. C. Harris and E. Raven, Green Chem., 2007, 9, 868–872 RSC.
- Y. C. Brito, V. M. Mello, C. C. S. Macedo, M. R. Meneghetti, P. A. Z. Suarez and S. M. P. Meneghetti, Appl. Catal., A, 2008, 351, 24–28 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |