Self-association based on orthogonal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O⋯CO interactions in the solid and liquid state†
O⋯CO interactions in the solid and liquid state†
Christoph
Fäh
,
Leo A.
Hardegger
,
Marc-Olivier
Ebert
,
W. Bernd
Schweizer
and
François
Diederich
*
Laboratorium für Organische Chemie, ETH Zürich, Hönggerberg, HCI, CH-8093 Zürich, Switzerland. E-mail: diederich@org.chem.ethz.ch; Fax: (+41)-44-632-1109
First published on 6th November 2009
Abstract
A network of orthogonal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯CO interactions was identified in the X-ray crystal structure of an α,α-difluorocyclopentanone derivative. This finding inspired investigations of self-association driven by these weak dipolar interactions in apolar solvents, which was proven by 1H NMR spectroscopy.
O⋯CO interactions was identified in the X-ray crystal structure of an α,α-difluorocyclopentanone derivative. This finding inspired investigations of self-association driven by these weak dipolar interactions in apolar solvents, which was proven by 1H NMR spectroscopy.
The early findings by Bolton that orthogonal C
O⋯CO contacts dominate the intermolecular interactions in the crystals of alloxan1a and triketoindane1b have been followed by numerous observations of such dipolar contacts in the crystal structures of small molecules, proteins and protein–ligand complexes.2 Allen et al. predicted the energy for the perpendicular interaction motif in a propanone dimer to be about a third (−7.6 kJ mol−1 at d(C⋯O) = 3.02 Å) of that of the rectangular antiparallel dipolar alignment (−22.3 kJ mol−1 at d(C⋯O) = 3.02 Å).3 Although weaker, the orthogonal CO⋯CO interaction in chemistry and biology predominates at closest contact distances, and this is a general finding for all dipolar interactions,2 presumably for steric reasons. Recently, the energetic contribution of intramolecular orthogonal CO⋯CO interactions has been quantified by applying a chemical double-mutant cycle to molecular torsion balances. In C6D6 at 298 K, the gain in free enthalpy due to this interaction was determined as −2.73 kJ mol−1 and high-level gas phase calculations using intermolecular perturbation theory (IMPT) revealed that attractive dispersion and electrostatic interactions are the major energetic contributors to the orthogonal dipolar association.4 Recent experimental data for geometrically constrained orthogonal CO⋯CO contacts between adjacent carbonyl groups in a polypeptide chain even provide evidence for the contribution of stabilising “Bürgi–Dunitz”-type5 n→π* interactions.6 Here, we present the first experimental evidence that two orthogonal CO⋯CO interactions are of sufficient strength to also stabilise intermolecular self-assembly in solution.
We recently reported the synthesis of α,α-difluorocyclopentanone and α,α-difluorocyclohexanone derivatives, which, in their hydrated form, act as inhibitors of the malarial aspartic protease Plasmepsin II.7 The X-ray crystal structure of (±)-1 (Fig. 1, P21/c) was solved, and the crystal lattice (Fig. 1 ESI†) revealed orthogonal CO⋯CO contacts through inversion centers at (0,0,0) and (0.5,0,0) to be the main intermolecular interactions between neighbouring enantiomeric pairs of molecules, generating two defined achiral dimer motifs.8 In type A dimer, the amide CO interacts with the CO of the difluoroketone moiety (d(C⋯O) = 3.05 Å, angle O⋯CO = 91°, angle CO⋯C = 161°), and in type B dimer, the CO of the difluoroketone points to the CO of the amide moiety (d(C⋯O) = 3.16 Å, angle O⋯CO = 92°, angle CO⋯C = 171°). Crystal packing seems to be dominated by the CO interaction as that part of the molecule shows surprisingly well oriented atoms compared to the heavily disordered pentyl and naphthyl fragments. The shorter contact is probably energetically the more favourable one, since the more nucleophilic amide CO interacts with the more electrophilic CO moiety of the difluoroketone. For crystal growth, (±)-1 was suspended in cyclohexane and heated to reflux. The resulting solution was cooled down to 22 °C and crystals of (±)-1 were isolated.
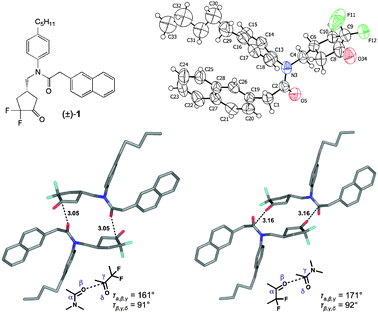 | ||
| Fig. 1 Top: The α,α-difluorocyclopentanone (±)-1 and its ORTEP representation. Thermal ellipsoids at 223 K are shown at the 50% probability level; for clarity only one position of the disordered pentyl group is shown. Bottom left: Type A dimer with (R2N)CO⋯C(CF2)O interactions between neighbouring molecules in the solid state. Bottom right: Type B dimer with C(CF2)O⋯(R2N)CO interactions between neighbouring molecules. Distances are given in Å. Colour code: C atoms, grey; O atoms, red; N atoms, blue; F atoms, light blue. | ||
We subsequently investigated whether these contacts are sufficiently strong to enable self-assembly in solution. Ketone (±)-1 was therefore dissolved in CD2Cl2 (dielectric constant ε = 8.93)9 and CDCl3 (ε = 4.89), but no substantial aggregation was detected by 1H NMR spectroscopy at 300 K, as no concentration-dependent changes in chemical shift were observed. Moving to less polar deuterated benzene (C6D6, ε = 2.27) on the other hand, concentration-dependent chemical shifts, indicative of self-association, were observed for (±)-1 at c = 0.001 M and c = 1.080 M (Fig. 2). This finding is in agreement with the results obtained with the unimolecular torsion balance system, for which a stronger CO⋯CO interaction was observed in C6D6 (ΔΔG = −2.73 ± 0.25 kJ mol−1) than in CD2Cl2 (ΔΔG = −1.22 ± 0.25 kJ mol−1) and CDCl3 (ΔΔG = −1.50 ± 0.25 kJ mol−1).4
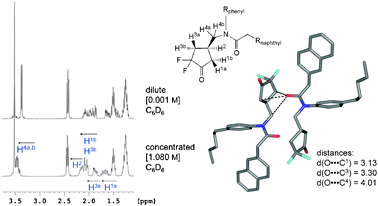 | ||
| Fig. 2 Left: Aliphatic region of the 1H NMR spectrum (400 MHz, 300 K) of (±)-1 in dilute and concentrated C6D6 solution. The spectra were referenced to the solvent signal. Right: The type A dimeric motif, based on two (R2N)CO⋯C(CF2)O interactions, from the X-ray crystal structure is analysed towards possible intermolecular influences of the amide carbonyl group on 1H NMR chemical shifts. | ||
The constitution and relative configuration of (±)-1 in C6D6 were confirmed by its 1H,1H COSY NMR spectrum (Fig. 2 ESI†), and the 1H NMR spectra at the two different concentrations subsequently analysed under the assumption that the type A dimeric assembly stabilised by the stronger COamide⋯COdifluoroketone interactions would prevail in solution (Fig. 2, Fig. 3 ESI†). The observed changes in chemical shift at the higher concentration of course result from the weighted average of all rapidly equilibrating aggregates, including those from homo- and heterochiral assembly; yet good support for the assumption of preferred type A dimerisation in solution was obtained.
The aromatic rings in (±)-1 are not directly involved in the dimeric assembly, yet downfield shifts of all phenyl resonances and upfield shifts of specific naphthyl signals are observed at the higher concentration (Fig. 3 ESI†). These shifts are indicative of a more efficient aromatic edge-to-face interaction in the dimers, resulting presumably from a reduction in conformational space in the self-assembled macrocycle. They are not indicative of any preferential self-association.
The changes in chemical shift observed for the aliphatic protons at the higher concentration, on the other hand, offer substantial support for the prevalence of the type A dimer, formed by the stronger (R2N)CO⋯C(CF2)O interactions (Fig. 2). The pentyl chain protons are not influenced upon increasing the concentration of (±)-1. On the other hand, the signals of the cyclopentyl ring protons show distinct changes in chemical shifts. Their concentration dependency can be fully explained by the formation of the type A dimer (Fig. 2). The largest Δδ-values are observed for protons H1a and H3a, which are strongly influenced by the deshielding effect of the nearby carbonyl oxygen of the binding partner in the dimer. Only small downfield shifts are observed for protons H1b, H2 and H3b. The protons H4a,b are not only deshielded by the carbonyl oxygen atom and therefore shifted downfield at the higher concentration, their resonance also undergoes a change in pattern, presumably reflecting the higher conformational rigidity of the dimeric assembly as compared to the monomer.
The observed Δδ-values do not support the presence of a significant amount of type B dimer, held together by the weaker C(CF2)O⋯(R2N)CO contacts, at the higher concentration (Fig. 4 ESI†). Firstly, the singlet of H5a,b would be strongly influenced by the carbonyl oxygen of the binding partner, which it is not, since no visible δ-change can be observed. Secondly, the protons H1a,b would be strongly influenced by the deshielding character of the carbonyl oxygen, but only the signal of H1a shifts substantially whereas H1b is only influenced slightly. And thirdly, protons H3a,b should not feel any nearby CO, but as discussed above, H3a appears strongly downfield shifted.
We subsequently undertook 1H NMR dilution studies (500 MHz, 300 K) to estimate the stability of this dimer. Curve fitting using IgorPro6 (for the equation, see ESI†)10 of the concentration-dependent change in chemical shift of three aliphatic (Fig. 3) and two aromatic (Fig. 5 ESI†) resonances yielded an average dissociation constant Kd = 1.48 M (association constant Ka = 0.68 M−1, ΔG = +0.98 kJ mol−1). Upon changing to perdeuterated cyclohexane (C6D12, ε = 2.02), competing interactions with solvent are further reduced and the 1H NMR dilution study (Fig. 6 ESI and 7 ESI†) provided a Kd-value of 0.057 M for the dimer (Ka = 17.54 M−1, ΔG = −7.1 kJ mol−1), which corresponds to a decrease in Kd by more than one order of magnitude, as compared to deuterated benzene.
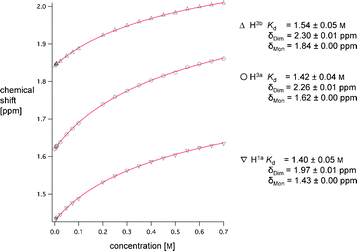 | ||
| Fig. 3 1H NMR dilution study (500 MHz, 300 K) of (±)-1 in C6D6 showing the shifts of the evaluated aliphatic resonances. The spectra were referenced to TMS. δDim is the chemical shift for the dimeric association at infinite concentration; δMon the chemical shift for monomer (±)-1 at infinite dilution. Curves were fitted with the software IgorPro6.10 | ||
Note that the changes in chemical shift induced by self-association in deuterated cyclohexane are smaller than those in benzene. This can be explained by the well established, larger influence of benzene solvation (“ASIS”, aromatic solvent-induced shifts)11 on the resonances of the monomers that exchange this solvation for dimeric aggregation.
Control experiments further confirmed that two CO⋯CO-interactions, and not aromatic interactions, are the major driving force for the observed self-association of (±)-1; they are described in detail in the ESI.† A tertiary N-methylamine S1 featuring the same aromatic substituents as (±)-1 did not show any significant self-association in the used solvent systems (Fig. 8 ESI†). The corresponding tertiary N-methylamide S2, also lacking the difluorocyclopentanone, displayed weak self-association, as judged from small observed changes in chemical shift during the NMR dilution study (Fig. 8 ESI†). In contrast, self-association becomes substantial when moving to (±)-S3 featuring the difluorocyclopentanone with a tertiary, purely aliphatic amide moiety – structurally similar to (±)-1 (Fig. 9 ESI†). Substantial changes in chemical shift are observed in dilution studies with this model compound and we presume a similar association geometry as proposed for (±)-1.
A similar self-assembly of two heterochiral monomers in C6D6 was also observed for the more rigid α,α-difluorocyclohexanone (±)-2 (ESI†).7 The changes in chemical shift observed in C6D6 upon moving from low to higher concentrations fully support the proposed dimeric self-association. Evaluation of a 1H NMR dilution experiment (500 MHz, 300 K) gave a Kd-value of 0.63 M for the dimer (Ka = 1.6 M−1, ΔG = −1.2 kJ mol−1) (Fig. 10 ESI†).
The reported thermodynamic data clearly represent estimates rather than highly reliable and accurate numbers, since solubility and, in C6D6, the weakness of association made it impossible to reach a high degree of self-association in the dilution study. Furthermore, we do not have direct evidence for the proposed dimeric association, although the curve fitting for this stoichiometry is very good. Vapour phase osmometric measurements were not successful in view of the weak association. In addition, an accurate measurement of diffusion coefficients was not possible which prevented the use of NMR diffusion experiments to elucidate the association stoichiometry (see ESI†).12 Nevertheless, the results demonstrate for the first time that orthogonal dipolar CO⋯CO interactions are a substantial intermolecular association force capable of inducing self-assembly in apolar, non-competing solvents. We are now developing tailor-made molecules for controlled self-assembly to further decipher and quantify the power of dipolar interactions in solution.
This work was supported by a grant from the ETH Research Council and a Novartis doctoral fellowship to C.F.
Notes and references
- (a) W. Bolton, Acta Crystallogr., 1964, 17, 147–152 CrossRef CAS; (b) W. Bolton, Acta Crystallogr., 1965, 18, 5–10 CrossRef CAS.
- R. Paulini, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 1788–1805 CrossRef CAS.
- F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320–329 CrossRef.
- F. R. Fischer, P. A. Wood, F. H. Allen and F. Diederich, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17290–17294 CrossRef CAS.
- H. B. Bürgi and J. D. Dunitz, Acc. Chem. Res., 1983, 16, 153–161 CrossRef.
- A. Choudhary, D. Gandla, G. R. Krow and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 7244–7246 CrossRef CAS.
- C. Fäh, L. A. Hardegger, L. Baitsch, W. B. Schweizer, S. Meyer, D. Bur and F. Diederich, Org. Biomol. Chem., 2009, 7, 3947–3957 RSC.
- C29H31F2NO2, MW = 463.57 g/mol, a = 6.1172(2) Å, b = 26.1410(10) Å, c = 15.5575(6) Å, α = 90.00°, β = 99.276(2)°, γ = 90.00°, V = 2455.3(2) Å3, 223 K, space group P21/c, Z = 4, independent reflections 6501 and 3364, respectively, final R(gt) = 0.1065, wR(gt) = 0.2838, CCDC 736727.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, 3rd edn, Wiley-VCH, Weinheim, 2003, pp. 472–475 Search PubMed.
- IgorPro6, Wavemetrics, Portland USA, 2009.
- F. Diederich, K. Dick and D. Griebel, Chem. Ber., 1985, 118, 3588–3619 CAS.
- (a) C. S. Johnson, Jr, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef; (b) Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data of (±)-1, reference compounds, additional data on the self-association studies in solution. CCDC 736727. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b912721f |
| This journal is © The Royal Society of Chemistry 2010 |