Polynitramino compounds outperform PETN†
Young-Hyuk Joo and Jean’ne M. Shreeve*
Department of Chemistry, University of Idaho, Moscow, ID 83844-2343, USA. E-mail: jshreeve@uidaho.edu; Fax: +1 208 885 9146; Tel: +1 208 885 6215
First published on 2nd November 2009
Abstract
New polynitramino compounds were synthesized and fully characterized using IR and multinuclear (1H, 13C, 15N) NMR spectroscopy, and elemental analysis as well as single-crystal X-ray diffraction.
Various energetic compounds have been investigated as precursors of high density energetic nitrimino,1 nitramino,2 nitro,3 or nitrato1a,3c,d compounds. In particular, high-energy polyazapolycyclic caged polynitramines have emerged as a promising family of high-energy density materials.2d The current well known polynitramino compounds are RDX, HMX and CL-20.
Primary nitramine molecules [dinitrourea (DNU),4 methylene dinitramine (MDNA)5 and ethylene dinitramine (EDNA)6] have impressive high densities, positive oxygen balances (except EDNA) and good detonation properties. However, the application of primary nitramines as energetic materials is limited due to their relatively low thermal stability (for DNU:4cTdec.: 92 °C, for MDNA:5Tdec.: 98–101 °C). Thermal decomposition studies involving aliphatic primary nitramines have led to an understanding of the relationship between structure and thermostability, and to the suggestion of a decomposition mechanism for these compounds.7a,b Increasing the number of nitro groups in a molecule in order to obtain a more balanced and powerful explosive inevitably results in an increase in acidity and a decrease in thermal stability of the resulting compounds.7
Here we describe the synthesis of a new energetic pentaerythrityl tetranitramine (PETNA) 4 that is a colorless crystal at ambient temperature, thermally decomposes at 183 °C, and has high density. Also described are the chemical, thermal, and sensitivity properties of 4, as well as some preliminary calculated detonation properties.
Pentaerythrityl tetranitramine (PETNA) 4 was prepared from pentaerythritol in overall good yields. Pentaerythrityl tetraurethane 2, prepared from aqueous tetraamine 18 and ethyl chloroformate, was nitrated using 100% nitric acid in trifluoroacetic anhydride to form tetranitrourethane 3. The latter was converted into 4 by ammonolysis with aqueous ammonia at 90 °C (Scheme 1). Within two hours, the ester groups were removed and the water-soluble ammonium salt was formed. After hydrolysis with concentrated hydrochloric acid, tetranitramine 4 was extracted with ethyl acetate and obtained in good yield. The structure of 4 was verified by single-crystal X-ray diffraction analysis (Fig. 1).‡ Efforts to nitrate urethane 2 did not yield the tetranitro derivative 3 when treated with only 100% nitric acid.
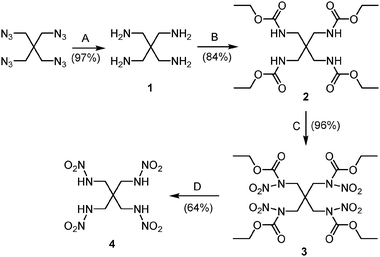 | ||
| Scheme 1 Synthesis of pentaerythrityl tetranitramine (PETNA) 4; A: H2–Pd/C, B: ClCOOCH2CH3, C: 100% HNO3–(CF3CO)2O, D: 28% aq. NH3–36% HCl. | ||
![Molecular structures (thermal ellipsoids represent 50% probability) of 4 (top) and 5 (bottom). Selected bond lengths [Å] and angles [°]: 4: C1–C2 1.5417(12), C2–N3 1.4598(16), N3–N4 1.3327(15), N4–O6 1.2263(15), N4–O5 1.2379(14), C1–C2–N3 114.39(9), C2–N3–N4 122.37(10); 5: C(1)–C(2) 1.5318(17), C2–N3 1.4463(18), N3–N4 1.3262(18), N4–O6 1.2230(17), N4–O5 1.2279(19), C1–C2–N3 113.74(12), C2–N3–N4 121.61(12).](/image/article/2010/CC/b916909a/b916909a-f1.gif) | ||
| Fig. 1 Molecular structures (thermal ellipsoids represent 50% probability) of 4 (top) and 5 (bottom). Selected bond lengths [Å] and angles [°]: 4: C1–C2 1.5417(12), C2–N3 1.4598(16), N3–N4 1.3327(15), N4–O6 1.2263(15), N4–O5 1.2379(14), C1–C2–N3 114.39(9), C2–N3–N4 122.37(10); 5: C(1)–C(2) 1.5318(17), C2–N3 1.4463(18), N3–N4 1.3262(18), N4–O6 1.2230(17), N4–O5 1.2279(19), C1–C2–N3 113.74(12), C2–N3–N4 121.61(12). | ||
Other successful syntheses of polynitramines 5 (77%), which was proved by single crystal X-ray diffraction analysis (Fig. 1), 6 (79%) and 7 (90%) by analogous nitration of polyurethane derivatives with concomitant retention of the pentaerythritol system resulted from extension of this reaction (Scheme 2).
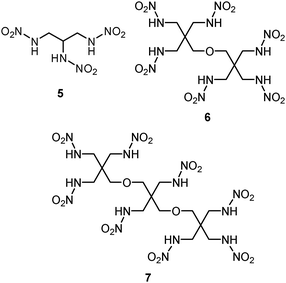 | ||
| Scheme 2 New polynitramines. | ||
The structures of all polynitramine derivatives 4–7 are supported by 1H, 13C and 15N NMR spectroscopic data (Table 1).
| a 1H, 13C and 15N NMR (CH3NO2 as external standard) spectra were recorded (using [D6]DMSO as solvent) at 300.1 MHz, 75.5 MHz and 50.7 MHz, respectively. The data for all compounds are summarized in the ESI.1 |
|---|
| 4: colorless crystal; 1H NMR: δ = 3.63 (s, 8H), 12.02 (s, 4H, NH); 13C NMR: δ = 42.2, 46.4; 15N NMR: δ = −207.6, −23.1. |
| 5: white crystal; 1H NMR: δ = 3.49 (dd, 2J = 14.7 Hz, 3J = 8.3 Hz, 2H), 3.74 (dd, 2J = 14.7 Hz, 3J = 4.5 Hz, 2H), 4.47 (m, 1H), 12.20 (s, 3H, NH); 13C NMR: δ = 44.4, 51.4; 15N NMR: δ = −202.1, −195.7, −19.6, −18.5. |
| 6: white crystal; 1H NMR: δ = 3.27 (s, 4H), 3.60 (s, 12H), 11.80 (br s, 6H, NH); 13C NMR: δ = 44.0, 46.1, 70.5; 15N NMR: δ = −202.3, −18.0. |
| 7: white solid; 1H NMR: δ = 3.26 (s, 4H), 3.60 (s, 16H), 11.94 (br s, 8H, NH); 13C NMR: δ = 43.7, 44.9, 45.7, 46.0, 70.5; 15N NMR: δ = −202.1, −201.9, −18.0, −17.8. |
The preparation of an asymmetric tetranitramine 11 was attempted by analogous nitration of the asymmetric tetraurethane 8 (Scheme 3; also see ESI†, Scheme S1). When the latter was nitrated with 100% nitric acid in trifluoroacetic anhydride, only a trinitro-substituted carbamate 9 was obtained as a colorless oil in a yield of 96%. Incomplete substitution of nitro groups results from steric hindrance of the three nitro urethane groups; this is significantly different from methylene bridged nitro urethane 3. The reaction of the asymmetric nitramine 10 with excess hydrazine hydrate in ethanol provided a further approach to the trihydrazinium salt (see ESI†).
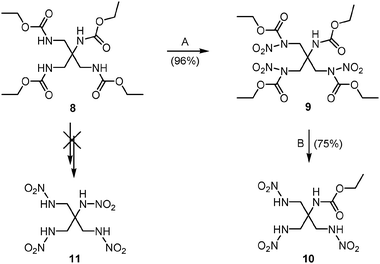 | ||
| Scheme 3 Attempt to synthesize asymmetric tetranitramine 11; A: 100% HNO3–(CF3CO)2O, B: 28% aq. NH3–36% HCl. | ||
The 15N NMR spectra9 of the polynitramines measured in [D6]DMSO show two or four resonances for nitrogen of NO2 and NH (see ESI†). In the spectra, a typical chemical shift for NO2 is observed between −23.1 and −17.8 ppm. The nitrogen signals from NH are found between −207.6 and −195.7 ppm at higher field than the starting material of polynitrourethane (between −167.8 and −173.0 ppm) and lower field than polyurethane with respect to the electronegativity effect (see 15N NMR spectra for polynitrourethane in the ESI†).
The thermal behavior of 4–7, which have decomposition temperatures between 181 and 185 °C (PETN: 160 °C, NG: 50–60 °C), was investigated using differential scanning calorimetry (Table 2). Only tris(nitramino)propane (5) shows a melting point (133 °C). Calculation of the heats of formation for polynitramines 4 and 5 was accomplished by using the Gaussian 03 suite of programs.10 The geometric optimizations of the structures and frequency analyses were carried out by using the B3LYP functional with the 6–31+G** basis set, and zero-point energies were calculated at the MP2/6–311++G** level.
| Compd | Tda/°C | Densityb/g cm−3 | ΔfH°298c/kJ mol−1 (kJ g−1) | Pd/GPa | De/m s−1 | ISf/J | OPg (%) |
|---|---|---|---|---|---|---|---|
| a Thermal decomposition temperature under nitrogen gas (DSC, 10 °C min−1).b Gas pycnometer (25 °C).c Heat of formation (using 83.68 kJ mol−1 for the enthalpy of sublimation for each compound; calculated via Gaussian 03).d Calculated detonation pressure (CHEETAH 5.0).e Calculated detonation velocity (CHEETAH 5.0).f Impact sensitivity (BAM Fallhammer).g OP = oxygen percent.h 5 has melting point at 133 °C.i Determined by X-ray crystallography (20 °C).j PETN = pentaerythrityl tetranitrate, RDX = 1,3,5-trinitro-1,3,5-triazacyclohexane, ref. 3c and 11.k NG = nitroglycerine, ref. 11 and 12. | |||||||
| 4 | 183 | 1.778 | −60.3 (−0.193) | 31.59 | 8657 | 6 | 41 |
| 5 | 183h | 1.753i | 324.9 (1.450) | 35.62 | 8933 | 20 | 43 |
| PETNj | 160 | 1.778 | −502.8 (−1.590) | 31.39 | 8564 | 2.9 | 61 |
| NGk | 50–60 | 1.60 | −351.5 (−1.548) | 25.3 | 7700 | 0.2 | 63 |
| RDXj | 230 | 1.816 | 92.6 (0.42) | 35.17 | 8977 | 7.4 | 43 |
All of the polynitramines exhibit negative heats of formation with 4 and 5 having the least negative −0.193 and 1.450 kJ g−1 (PETN: −1.590 kJ g−1, NG: −1.548 kJ g−1). The detonation parameters [pressures (P) and velocities (D)] were calculated using the CHEETAH 5.0 computer program.13 The program is based on the traditional Chapman–Jouget thermodynamic detonation theory. For 4 and 5 the calculated detonation pressures are 31.59 and 35.62 GPa (PETN: 31.39 GPa, NG: 25.3 GPa). Detonation velocities are 8657 and 8933 m s−1, respectively (PETN: 8564 m s−1, NG: 7700 m s−1). The impact sensitivity was tested according to BAM methods (BAM Fallhammer).14 In Table 1, there is a range in impact sensitivities from the sensitive 4 (6 J) to 5 (20 J) and finally to the insensitive compound 6 (>40 J) and 7 (>40 J). The sensitivity of these polynitramino compounds is greatly reduced compared to PETN (2.9 J) and NG (0.2 J).
Safety precautions: while we have experienced no difficulties with the shock instability of polyazide and polynitramino compounds, manipulations must be carried out in a hood behind a safety shield. Leather gloves must be worn.
The authors gratefully acknowledge the support of DTRA (HDTRA1-07-1-0024), NSF (CHE-0315275), and ONR (N00014-06-1-1032). We are grateful to Dr D. A. Parrish, Naval Research Laboratory (NRL), for determining the single-crystal X-ray structures.
Notes and references
- (a) Y.-H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2009, 48, 564 CrossRef CAS; (b) Y.-H. Joo and J. M. Shreeve, Chem.–Eur. J., 2009, 15, 3198 CrossRef CAS; (c) J. Stierstorfer, K. R. Tarantik and T. M. Klapötke, Chem.–Eur. J., 2009, 14, 5775 CrossRef; (d) H. Gao, Y. Huang, C. Ye, B. Twamley and J. M. Shreeve, Chem.–Eur. J., 2008, 14, 5596 CrossRef CAS; (e) T. M. Klapötke, J. Stierstorfer and A. U. Wallek, Chem. Mater., 2008, 20, 4519 CrossRef; (f) H. Xue, H. Gao, B. Twamley and J. M. Shreeve, Chem. Mater., 2007, 19, 1731 CrossRef CAS.
- (a) R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584 CrossRef CAS; (b) G. Steinhauser and T. M. Klapötke, Angew. Chem., Int. Ed., 2008, 47, 3330 CrossRef; (c) R. P. Singh, H. Gao, D. T. Meshri and J. M. Shreeve, in High Energy Density Materials, ed. T. M. Klapötke, Springer, Berlin, Heidelberg, 2007, vol. 37 Search PubMed; (d) T. M. Klapötke, in High Energy Density Materials, ed. T. M. Klapötke, Springer, Berlin, Heidelberg, 2007, p. 85 Search PubMed.
- (a) M. Göbel and T. M. Klapötke, Adv. Funct. Mater., 2009, 19, 347 CrossRef; (b) Y. Huang, H. Gao, B. Twamley and J. M. Shreeve, Chem.–Eur. J., 2009, 15, 917 CrossRef CAS; (c) D. E. Chavez, M. A. Hiskey, D. L. Naud and D. Parrish, Angew. Chem., Int. Ed., 2008, 47, 8307 CrossRef CAS; (d) W.-G. Liu, S. V. Zybin, S. Dasgupta, T. M. Klapötke and W. A. Goddard, J. Am. Chem. Soc., 2009, 131, 7490 CrossRef CAS; (e) T. M. Klapötke, P. Mayer, C. M. Sabate, J. M. Welch and N. Wiegand, Inorg. Chem., 2008, 47, 6014 CrossRef; (f) T. M. Klapötke, C. M. Sabate and M. Rasp, Dalton Trans., 2009, 1825 RSC; (g) M. Göbel, B. H. Tchitchanov, J. S. Murray, P. Politzer and T. M. Klapötke, Nat. Chem., 2009, 1, 229 Search PubMed.
- (a) P. Goede, N. Wingborg, H. Bergman and N. V. Latypov, Propellants, Explos., Pyrotech., 2001, 26, 17 CrossRef CAS; (b) C. Ye, H. Gao, B. Twamley and J. M. Shreeve, New J. Chem., 2008, 32, 317 RSC; (c) D. B. Lempert and I. N. Zyuzin, Propellants, Explos., Pyrotech., 2007, 32, 360 CrossRef CAS.
- R. C. Brian and A. H. Lamberton, J. Chem. Soc., 1949, 1633 RSC.
- W. E. Bachmann, W. J. Horton, E. L. Jenner, N. W. MacNaughton and C. E. Maxwell III, J. Am. Chem. Soc., 1950, 72, 3132 CrossRef CAS.
- (a) R. S. Stepanov, A. M. Astachov and L. A. Kruglyakova, 29th International Annual ICT-Conference, 1998, 128/1–128/7 Search PubMed; (b) A. M. Astachov, R. R. Stepanov, L. A. Kruglyakova and Y. V. Kekin, 31th International Annual ICT-Conference, 2000, 13/1–13/10 Search PubMed; (c) Y. Lee, P. Goede, N. Latypov and H. Östmark, 36th International Annual ICT-Conference, 2005, 124/1–124/9 Search PubMed.
- W. Hayes, H. M. Osborn, S. D. Osborne, R. A. Rastall and B. Romagnoli, Tetrahedron, 2003, 59, 7983 CrossRef CAS , the method of hydrogenation of azide was modified. See ESI†.
- S. Berger, S. Braun and H.-O. Kalinowski, NMR-Spektroskopie von Nichtmetallen, Band 2, 15N-NMR-Spektroskopie, Georg Thieme Verlag, Stuttgart, New York, 1992, pp. 36 Search PubMed.
- M. J. Frisch et al., GAUSSIAN 03 (Revision D.01), Search PubMedsee ESI†.
- (a) H. H. Klause, in Energetic Materials, ed. U. Teipel, VCH, Weinheim, 2005, p. 1 Search PubMed; (b) R. Meyer, J. Köhler and A. Homburg, in Explosives, Wiley-VCH, Weinheim, 2007 Search PubMed.
- T. M. Klapötke, A. Penger, S. Scheutzow and L. Vejs, Z. Anorg. Allg. Chem., 2008, 634, 2994 CrossRef.
- S. Bastea, L. E. Fried, K. R. Glaesemann, W. M. Howard, P. C. Souers and P. A. Vitello, CHEETAH 5.0 User’s Manual, Lawrence Livermore National Laboratory, 2007 Search PubMed.
- (a) www.bam.de; (b) A portion of polynitramine (20 mg) was subjected to a drop-hammer test using a 5 or 10 kg weight dropped. (Insensitive > 40 J; less sensitive ≥35 J; sensitive ≥4 J; very sensitive ≤3 J).
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental procedures, analytical data, and NMR spectra of products. CCDC 737611 (4) and 737612 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b916909a |
‡ Crystallographic data compound 4: C5H12N8O8, Mr = 312.23, tetragonal, I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) , a = b = 9.6543(11) Å, c = 6.3283(9) Å, α = β = γ = 90°, V = 589.83(13) Å3, Z = 2, ρcalcd = 1.758 g cm−3, T = 293(2) K, μ = 0.163 mm−1, F(000) = 324, R1 = 0.0256 for 610 observed (I > 2σI) reflections and 0.0258 for all 615 reflections (Rint = 0.0180), goodness-of-fit = 1.113, 48 parameters. 5: C3H8N6O6, Mr = 224.15, orthorhombic, Pnma, a = 6.7812(7) Å, b = 13.9258(13) Å, c = 8.9932(9) Å, α = β = γ = 90°, V = 849.26(15) Å3, Z = 4, ρcalcd = 1.753 g m−3, T = 293(2) K μ = 0.166 mm−1, F(000) = 464, R1 = 0.0354 for 808 observed (I > 2σI) reflections and 0.0394 for all 911 reflections (Rint = 0.0169), goodness-of-fit = 1.069, 76 parameters. , a = b = 9.6543(11) Å, c = 6.3283(9) Å, α = β = γ = 90°, V = 589.83(13) Å3, Z = 2, ρcalcd = 1.758 g cm−3, T = 293(2) K, μ = 0.163 mm−1, F(000) = 324, R1 = 0.0256 for 610 observed (I > 2σI) reflections and 0.0258 for all 615 reflections (Rint = 0.0180), goodness-of-fit = 1.113, 48 parameters. 5: C3H8N6O6, Mr = 224.15, orthorhombic, Pnma, a = 6.7812(7) Å, b = 13.9258(13) Å, c = 8.9932(9) Å, α = β = γ = 90°, V = 849.26(15) Å3, Z = 4, ρcalcd = 1.753 g m−3, T = 293(2) K μ = 0.166 mm−1, F(000) = 464, R1 = 0.0354 for 808 observed (I > 2σI) reflections and 0.0394 for all 911 reflections (Rint = 0.0169), goodness-of-fit = 1.069, 76 parameters. |
| This journal is © The Royal Society of Chemistry 2010 |