An enzymatic route to 5-deoxy-5-[18F]fluoro-D-ribose, a [18F]-fluorinated sugar for PET imaging†
Mayca
Onega
ab,
Juozas
Domarkas
b,
Hai
Deng
c,
Lutz F.
Schweiger
b,
Timothy A. D.
Smith
b,
Andrew E.
Welch
b,
Christophe
Plisson
d,
Antony D.
Gee
d and
David
O’Hagan
*a
aUniversity of St Andrews, School of Chemistry and Centre for Biomolecular Sciences, North Haugh, St Andrews, Fife, UK KY16 9ST. E-mail: do1@st-andrews.ac.uk; Fax: +44 01334 463800; Tel: +44 01334 467171
bJohn Mallard Scottish PET Centre, School of Medical Sciences, University of Aberdeen, Foresterhill, Aberdeen, UK AB25 2ZD
cUK Marine Biodiscovery Centre, Department of Chemistry, Meston Walk, University of Aberdeen, Aberdeen, UK AB24 3UE
dGSK Clinical Imaging Centre, Imperial College London, Hammersmith Hospital, Du Cane Road, London, UK W12 0NN
First published on 10th November 2009
Abstract
An efficient two-step, one-pot, biotransformation involving the fluorinase enzyme is described for the synthesis of 5-deoxy-5-[18F]fluororibose, a novel [18F]-fluorinated sugar suitable for positron emission tomography (PET) applications.
Positron emission tomography (PET) is a powerful imaging technique which is rapidly growing in importance in diagnosis particularly as a method for imaging tumours (oncology), brain defects (neurology) and in the arena of clinical trials for monitoring the distribution and clearance of drugs in the body. 2-Deoxy-2-[18F]fluoroglucose ([18F]FDG) is the most widely used tool in PET studies and administration of [18F]FDG constitutes about 90% of all PET investigations in the clinic.1,2 The fluorine-18 isotope is attractive because of its relatively long half-life (110 min) compared to 11C (20 min), 13N (10 min), 15O (2 min), respectively for the other most common isotopes used to label ligands or metabolites for PET imaging. A current challenge for 18F-labelled radiopharmaceuticals is to develop new chemistry for rapid and clean synthesis and with straightforward purification protocols for new in vivo imaging probes.2,3 We have been exploring an enzymatic method for 18F–C bond formation,4 although generally enzymatic syntheses for PET are unusual due to the virtual absence of appropriate biocatalysts.5 However, the identification and over-expression of a fluorination enzyme (fluorinase) from the bacterium Streptomyces cattleya6 has opened up a new approach for the selective incorporation of organic fluorine from fluoride ion in PET syntheses.2–4 The fluorinase (5′-fluoro-5′-deoxyadenosine synthase, 5′-FDAS, E.C. 2.5.1.63) catalyses the conversion of S-adenosyl-L-methionine 1 (SAM) and fluoride ion to 5′-fluoro-5′-deoxyadenosine 2 (5′-FDA) and L-methionine 3 (Scheme 1). 7
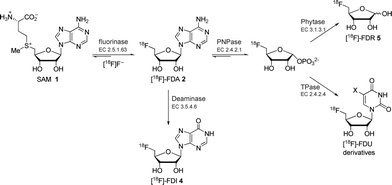 | ||
| Scheme 1 Previously reported fluorinase coupled enzyme systems for the synthesis of various 18F-labelled compounds.8 | ||
We have previously reported a series of fluorinase coupled enzyme transformations starting from [18F]fluoride ion and generating 5′-[18F]FDA 2, 5′-deoxy-5′-[18F]fluoroinosine 4 (5′-[18F]FDI) and 5-deoxy-5-[18F]fluoro-D-ribose 5 (5-[18F]FDR) in very good radiochemical incorporations (RCIs) and within a few hours.8 The biotransformation routes to these molecules are shown in Scheme 1.
More recently, a series of fluorinase coupled base swap biotransformations were demonstrated to prepare a range of 5′-deoxy-5′-[18F]fluoro-nucleosides, by coupling the fluorinase to purine and pyrimidine nucleotide phosphorylases, and thus the portfolio of 18F-labelled compounds is being extended.9 In this Communication a significantly improved synthesis of 5-[18F]FDR 5 is reported. The previously reported synthesis of 5-[18F]FDR 5 involved a three-enzyme system consisting of the fluorinase, an immobilised purine nucleotide phosphorylase (PNPase) and a phytase (Scheme 1). This resulted in the production of 5-[18F]FDR 5 reaching a RCI of 45% after 4 h.8 In view of the prominent role of the monosaccharide [18F]FDG in PET technology,1–3 we decided to try to optimise the synthesis of 5-[18F]FDR 5 to explore the potential of this monosaccharide ribose in PET imaging. We have been able to reduce the synthesis from a three to a two enzyme step protocol and with a much improved radiochemical incorporation. To date the coupled enzyme reactions have utilised a PNPase to remove the purine base and generate ribose phosphate (Scheme 1). These enzymes are involved in the usual metabolism of purines. Nucleoside hydrolases (NHs, E.C. 3.2.2.-), which utilise water rather than phosphate as a nucleophile, are less widely distributed in nature and their physiological role is only well understood in certain parasitic protozoa (e.g., Trypanosoma).10 In these pathogenic organisms, NHs play a vital role in the purine salvage pathway, catalysing the hydrolysis of the assimilated nucleosides and allowing the recycling of the purine bases and ribose. The nucleoside hydrolase from Trypanosoma vivax (TvNH) has been well characterised structurally and kinetically. This NH catalyses the hydrolysis of the N-glycosidic bond of inosine, adenosine and guanosine most efficiently (Scheme 2).11
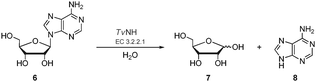 | ||
| Scheme 2 The NH-catalysed hydrolysis of a ribonucleoside exemplified by adenosine 6N-glycosidic bond cleavage to generate ribose 7 and adenine 8. | ||
It became an objective to explore the recently cloned and over-expressed TvNH in a biotransformation to generate 5-FDR 5. Coupling the fluorinase to the TvNH offered an attractive strategy for a two-step preparation of 5-[18F]FDR 5 (Scheme 3).
![Two-step biotransformation for the synthesis of 5-[18F]FDR 5 starting from SAM 1.](/image/article/2010/CC/b919364b/b919364b-s3.gif) | ||
| Scheme 3 Two-step biotransformation for the synthesis of 5-[18F]FDR 5 starting from SAM 1. | ||
Recombinant TvNH was purified from extracts of E. coli strain WK6, transformed with the pQE30-IAGNH plasmid, giving yields of about 30 mg protein/L medium.12 The next step in the study involved exploring the combination of these two enzymes for the radiolabelled synthesis of 5-[18F]FDR 5. Initially, a one-pot reaction was explored but this proved slow and inefficient, and trial and error led to a much more efficient sequential one-pot reaction with an intermediate heating protocol to precipitate the step one enzymes (fluorinase/L-AAO). For that, the fluorinase/L-AAO was incubated with SAM 1 in the presence of [18F]fluoride ion at 37 °C and pH 7.0, as previously reported.8 HPLC analysis of the reaction mixture after 1 h showed a 97% RCI (decay corrected) of 5-[18F]FDA 2 from [18F]fluoride ion. The fluorinase/L-AAO was then heat denatured (95 °C) and the protein precipitated, and then TvNH was added directly to a final concentration of 20 mg/mL, in HEPES buffer (pH 8.0). Incubation was then extended for a further 2 h and the reaction was followed by radio-HPLC analysis (Fig. 1a). After this time, formation of 5-[18F]FDR 5 from 5-[18F]FDA 2 was approximately 80% RCI with decay correction (Fig. 1b). This approach has significantly shortened reaction times and improved the radiochemical conversion of 5-[18F]FDR 5 relative to the previously reported three-enzyme synthesis.8 Although the reactions take 1–3 hours, rather than minutes, the half life of fluorine-18 is sufficiently long that this does not present a major problem for imaging studies.
![(a) Time course (in triplicate) showing the second phase of the biotransformation indicating the production of 5-[18F]FDR 5 from 5′-[18F]FDA 2 catalysed by TvNH at 37 °C, pH 8.0. (b) Radiogram showing the product profile of the sequential one-pot biotransformation illustrating ∼80% radiochemical incorporation of 5-[18F]FDR 5 from [18F]fluoride ion after a 3 h incubation.](/image/article/2010/CC/b919364b/b919364b-f1.gif) | ||
| Fig. 1 (a) Time course (in triplicate) showing the second phase of the biotransformation indicating the production of 5-[18F]FDR 5 from 5′-[18F]FDA 2 catalysed by TvNH at 37 °C, pH 8.0. (b) Radiogram showing the product profile of the sequential one-pot biotransformation illustrating ∼80% radiochemical incorporation of 5-[18F]FDR 5 from [18F]fluoride ion after a 3 h incubation. | ||
Preliminary cell uptake studies with two human breast cancer cell lines (MDA-MB-468 and MDA-MB-453) show good uptake of 5-[18F]FDR 5 and very limited metabolism over a 2 h period (HPLC-radiochemical detection). Small animal imaging studies in small animal models are now underway.
In summary, an efficient synthesis of 5-deoxy-5-[18F]fluoro-D-ribose 5 from [18F]fluoride ion has been achieved via a novel enzymatic approach in which the fluorinase from S. cattleya is combined with a nucleoside hydrolase in a sequential one-pot reaction. The experiments demonstrate a two-enzyme route to 5-[18F]FDR 5 in good radiochemical incorporation. The fluorinase enzyme is a stable enzyme and the present study further extends the range and versatility of the fluorinase as a biocatalyst for the efficient synthesis of 5-[18F]FDR 5, which makes it available for cell uptake studies and small animal model imaging.
We are grateful for Grants from the Medical Research Council (GO802567) and BBSRC (BB/F007426/1) for financial support. We are also indebted to Prof. W. Versées, Prof. J. Steyaert and Dr J. Barlow (Vrije Universiteit Brussel, Belgium) for providing a recombinant E. coli strain containing the nucleoside hydrolase gene from T. vivax. Mr Stuart Craib (Aberdeen University) is acknowledged for fluorine-18 production.
Notes and references
- (a) J. Czernin, H. R. Schelbert, D. H. S. Silverman and W. P. Melega, in PET: Molecular Imaging and its Biological Applications, ed. M. E. Phelps, Springer-Verlag, Berlin, Heidelberg, New York, 2nd edn, 2004, vol. 1, ch. 4, pp. 321–584 Search PubMed; (b) Positron Emission Tomography: Basic Sciences, ed. D. L. Bailey, D. W. Townsend, P. E. Valk and M. N. Maisey, Springer-Verlag, London, 2005 Search PubMed; (c) G. Muehllehner and J. S. Karp, Phys. Med. Biol., 2006, 51, 117–137.
- Fluorine and Health—Molecular Imaging: Biomedical Materials and Pharmaceuticals, ed. A. Tressaud and G. Haufe, Elsevier, Amsterdam, Oxford, 2008 Search PubMed.
- (a) L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2008, 2853–2873 CrossRef CAS; (b) M.-C. Lasne, C. Perrio, J. Rouden, L. Barré, D. Roeda, F. Dolle and C. Crouzel, in Chemistry of β+-Emitting Compounds Based on Fluorine-18, in Topics in Current Chemistry, Springer-Verlag, Berlin, Heidelberg, 2002, vol. 2, pp. 201–258 Search PubMed.
- L. Martarello, Schaffrath, H. Deng, A. D. Gee, A. Lockhart and D. O’Hagan, J. Labelled Compd. Radiopharm., 2003, 46, 1–9 CrossRef.
- (a) P. Bjurling, Y. Watanabe, M. Tokushige, T. Oda and B. Långström, J. Chem. Soc., Perkin Trans. 1, 1989, 1331–1334 RSC; (b) P. Bjurling, G. Antoni, Y. Watanabe and B. Långström, Acta Chem. Scand., 1990, 44, 178–182 CrossRef CAS; (c) E. Lui, R. Chirakal and G. Firnau, J. Labelled Compd. Radiopharm., 1998, 41, 503–521 CrossRef CAS; (d) G. Antoni, H. Omura, M. Ikemoto, R. Moulder, Y. Watanabe and B. Långström, J. Labelled Compd. Radiopharm., 2001, 44, 287–294 CrossRef CAS.
- (a) D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef CAS; (b) C. Schaffrath, H. Deng and D. O’Hagan, FEBS Lett., 2003, 547, 111–114 CrossRef CAS; (c) C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O’Hagan and J. H. Naismith, Nature, 2004, 427, 561–565 CrossRef CAS.
- C. D. Cadicamo, J. Courtieu, H. Deng, A. Meddour and D. O’Hagan, ChemBioChem, 2004, 5, 685–690 CrossRef.
- (a) H. Deng, S. L. Cobb, A. D. Gee, A. Lockhart, L. Martarello, R. P. McGlinchey, D. O’Hagan and M. Onega, Chem. Commun., 2006, 652–654 RSC; (b) M. Onega, M. Winkler and D. O'Hagan, Future Med. Chem., 2009, 1, 865–873 Search PubMed.
- M. Winkler, J. Domarkas, L. F. Schweiger and D. O’Hagan, Angew. Chem., Int. Ed., 2008, 47, 10141–10143 CrossRef CAS.
- (a) W. Versées and J. Steyaert, Curr. Opin. Struct. Biol., 2003, 13, 731–738 CrossRef CAS; (b) E. Iovane, B. Giabbai, L. Muzzolini, V. Matafora, A. Fornili, C. Minici, F. Giannese and M. Degano, Biochemistry, 2008, 47, 4418–4426 CrossRef CAS; (c) D. J. Hammond and W. E. Gutteridge, Mol. Biochem. Parasitol., 1984, 13, 243–261 CrossRef CAS.
- (a) W. Versées, K. Decanniere, R. Pellé, J. Depoorter, E. Brosens, D. W. Parkin and J. Steyaert, J. Mol. Biol., 2001, 307, 1363–1379 CrossRef CAS; (b) D. Mazumder-Shivakumar and T. C. Bruice, Biochemistry, 2005, 44, 7805–7817 CrossRef CAS; (c) G. Huysmans, A. Ranquin, L. Wyns, J. Steyaert and P. Van Gelder, J. Controlled Release, 2005, 102, 171–179 CrossRef CAS; (d) A. Vandemeulebroucke, W. Versées, J. Steyaert and J. N. Barlow, Biochemistry, 2006, 45, 9307–9318 CrossRef CAS; (e) W. Versées, J. Barlow and J. Steyaert, J. Mol. Biol., 2006, 359, 331–346 CrossRef CAS; (f) A. Vandemeulebroucke, S. De Vos, E. Van Holsbeke, J. Steyaert and W. Versées, J. Biol. Chem., 2008, 283, 22272–22282 CrossRef CAS; (g) W. Versées, A. Goeminne, M. Berg, A. Vandemeulebroucke, A. Haemers, K. Augustyns and J. Steyaert, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 953–960 CrossRef CAS.
- A TvNH E. coli over-expression system was a kind gift from Prof. W. Versées, Prof. J. Steyaert and Dr J. Barlow, Vrije Universiteit Brussel, Belgium. 5′-FDA 2 is a relatively poor substrate for TvNH (KM ≈ 1.2 mM, kcat ≈ 0.01 s−1); personal communication, Prof. W. Versées, Vrije Universiteit Brussel, Belgium.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b919364b |
| This journal is © The Royal Society of Chemistry 2010 |