Synthesis and photophysical properties of a hydrogen-bonded phthalocyanine–perylenediimide assembly†
Wolfgang
Seitz
a,
Ángel J.
Jiménez
b,
Esther
Carbonell
a,
Bruno
Grimm
a,
M. Salomé
Rodríguez-Morgade
b,
Dirk M.
Guldi
*a and
Tomás
Torres
*b
aFriedrich-Alexander-Universität Erlangen-Nürnberg, Institute for Physical Chemistry, Egerlandstr. 3, D-91058 Erlangen, Germany. E-mail: dirk.guldi@chemie.uni-erlangen.de
bUniversidad Autónoma de Madrid, Departamento de Química Orgánica, Campus de Cantoblanco, 28049-Madrid, Spain. E-mail: tomas.torres@uam.es
First published on 19th November 2009
Abstract
A supramolecular phthalocyanine–perylenediimide donor–acceptor array has been assembled by using a melamine/perylenediimide motif. Photoexcitation of the perylenediimide component affords transduction of singlet excited state energy to the energetically lower lying phthalocyanine.
Multichromophoric arrays able to harvest visible light photons and produce energy and/or electron transfer processes are essential targets to construct artificial photosynthetic devices for photovoltaic and optoelectronic applications.1 Owing to the excellent light harvesting features of phthalocyanines (Pcs), especially when compared to other types of porphyrinoids, they have emerged as superior materials for solar energy conversion applications.2,3 Thus, they display high molar absorption coefficients with an absorption maximum at the highest solar flux, namely in the range of 700 nm.4 Establishing versatile protocols to (i) vary the central metal and (ii) alter the peripheral functionalization has led to full control over the relationship between energy and charge transfer dynamics in multichromophoric arrays.5–7 A key asset of phthalocyanines is their small reorganization energies so that they give rise to charge transfer reactions, which consequently evokes an acceleration of the charge separation step, while the energy wasting and undesired charge recombination is subject to a notable deceleration.8–10 All these beneficial features have led us to use phthalocyanines in different electron donor–acceptor designs.7,8,11
Yet it has not been until very recently that perylenediimides (PDI)12 have been probed as the oxidizing complement to phthalocyanines en route toward ensembles producing energy transfer and long-lived radical ion pair states.6–9,13 In our previous work, we focused on the implementation of phthalocyanines and perylenediimides via the synthesis of two phthalocyanine–perylenediimide electron donor–acceptor conjugates/hybrids. In the hybrid structure, the assembly was realized through metal coordination at the imido positions of the PDI.7 The conjugate, on the other hand, was based on two Pcs that were connected to the PDI bay region by triple bonds.8 In the resulting assemblies efficient charge transfer evolved from Pc to PDI to afford in both cases radical ion pair states with lifetimes in the nanosecond scale.
The design of noncovalent donor–acceptor systems has been shown to be a promising strategy for fundamental charge-transfer studies.12,14 Complementary hydrogen bonding interactions are especially suitable for the assembly of well-defined nanostructures, due to their specificity and directionality. Inspired by the pioneering work on the melamine/barbituric acid motif,15 the melamine/PDI motif has been systematically pursued to arrange two- or three-dimensional nanostructures with well-defined geometries.12,16,17 Following our interest in the basic understanding of the donor–acceptor properties of phthalocyanines in multicomponent systems focusing on the preparation of solar cells,3 herein we wish to report the functionalization of a phthalocyanine with a ditopic melamine moiety, which results in 5 (Fig. 1) that binds to a complementary bifunctional PDI 1 (Fig. 1) by means of triple hydrogen bonding, to afford the corresponding Pc–PDI supramolecular system. Photophysical studies on the assembly are also carried out. The perylenediimide derivative 1 (Fig. 1) was prepared following reported procedures.17 The melamine-substituted phthalocyanine 5 was synthesised in moderate yields by Sonogashira coupling of the corresponding 2,4-dioctylamino-6-ethynyl-1,3,5-triazine (3) and the iodo-substituted phthalocyanine 4 (see supporting information, Scheme 1†).18
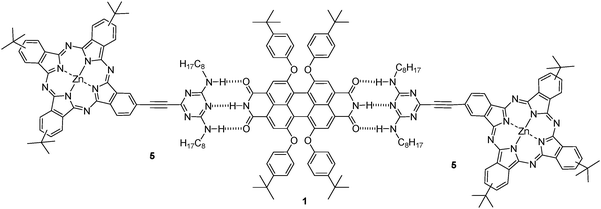 | ||
| Fig. 1 Pc–PDI supramolecular assembly. | ||
For both PDI 1 and ZnPc 5, strong ground state absorption features are seen in the visible range of the solar spectrum (Fig. 2). In particular, for 1 the characteristic 0–*2, 0–*1, and 0–*0 transitions evolve in the 400 to 600 nm range with transitions of increasing oscillator strength at 440 nm < 526 nm < 565 nm in THF. ZnPc 5, on the other hand, reveals the corresponding 0–*2, 0–*1, and 0–*0 transitions at 352, 610/630, and 670/686 nm, again with increasing oscillator strength in THF.19 The large splitting of the Q-band (16 nm) reflects the reduction of symmetry upon introduction of the triazine conjugated to the macrocycle. Notable are the complementary absorptions of PDI 1 and ZnPc 5, that is, absorbing in different parts of the solar spectrum.
![Upper part: absorption spectra of a dilute benzonitrile solution of PDI 1 (3.0 × 10−6 M) with variable concentrations of ZnPc 5 (0; 2.97 × 10−7; 8.74 × 10−7; 1.43 × 10−6; 2.22 × 10−6; 2.97 × 10−6; 3.91 × 10−6; 4.79 × 10−6; 5.61 × 10−6; 6.00 × 10−6; 6.92 × 10−6; 8.02 × 10−6; 9.02 × 10−6; 1.01 × 10−5; 1.11 × 10−5; 1.20 × 10−5; 1.31 × 10−5; 1.40 × 10−5; 1.50 × 10−5 M). Arrows indicate the progression of the titration. Central part: emission spectra upon 526 nm excitation. Lower part: I/I0 of PDI 1versus [ZnPc] relationship used to determine the association constant.](/image/article/2010/CC/b921363e/b921363e-f2.gif) | ||
| Fig. 2 Upper part: absorption spectra of a dilute benzonitrile solution of PDI 1 (3.0 × 10−6 M) with variable concentrations of ZnPc 5 (0; 2.97 × 10−7; 8.74 × 10−7; 1.43 × 10−6; 2.22 × 10−6; 2.97 × 10−6; 3.91 × 10−6; 4.79 × 10−6; 5.61 × 10−6; 6.00 × 10−6; 6.92 × 10−6; 8.02 × 10−6; 9.02 × 10−6; 1.01 × 10−5; 1.11 × 10−5; 1.20 × 10−5; 1.31 × 10−5; 1.40 × 10−5; 1.50 × 10−5 M). Arrows indicate the progression of the titration. Central part: emission spectra upon 526 nm excitation. Lower part: I/I0 of PDI 1versus [ZnPc] relationship used to determine the association constant. | ||
Fluorescence spectra which were recorded for 1 and 5 mirror image the aforementioned absorptions. In THF, 593 and 650 nm are the *0–0 and *0–1 maxima for PDI 1, while for ZnPc 5 the *0–0 maximum is seen at 692 nm. The quantum yields are 1 and 0.31 for 1 and 5, respectively.12 Details on forming PDI/ZnPc came from different experiments. The optical spectrum of Pc 5 in chloroform displays a broad Q-band with shoulders at 740 and 840 nm, that suggest the presence of aggregated species in solution. Upon addition of the PDI 1, the Q-band sharpens with concomitant reduction of the aggregation bands. The effect is particularly noticeable when the Pc/PDI ratio is 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (see Fig. S-8†).20 Moreover, dilute solutions of PDI 1 (3.0 × 10−6 M)—in THF or benzonitrile (Fig. 2)—were titrated with variable amounts of ZnPc 5 (0–1.50 × 10−5 M) and probed by means of absorption and fluorescence spectroscopies. A 1:1 mixture in THF, for example, displays PDI centered (434, 527, and 568 nm) and ZnPc centered transitions (609, 632, 670, and 686 nm) in approximately a 1:3 ratio—identical with the simple superimposition of PDI 1 and ZnPc 5. Still, the presence of isosbestic points indicates the transformation of 1 (i.e., starting-point of the titration) into PDI/ZnPc (i.e., end-point of the titration). Less ambiguous were the corresponding excitation experiments following 526 nm excitation. Here, the PDI fluorescence, which evolves in the blue with a quantum yield of 1, decreased exponentially in the presence of variable concentrations of ZnPc 5. All throughout the titration, the PDI fluorescence is subject to a 60% quenching in THF, while in benzonitrile the overall quenching amounts to 40%. The exponential concentration/fluorescence relationship enabled the affinity constant, corresponding to the formation of PDI/ZnPc from PDI 1 and ZnPc 5, to be calculated. In THF and benzonitrile, the binding constants are 2 × 105 M−1 and 7 × 104 M−1, respectively. The fluorescence decrease is accompanied by a gradual blue shift of the main fluorescence band from 593 to 588 nm in THF and from 612 to 601 nm in benzonitrile. In the red part of the spectrum, namely around 700 nm, we see the characteristic ZnPc fluorescence emerging. The quantum yield of the latter—formed indirectly—is, however, with 0.063 (THF) and 0.074 (benzonitrile) markedly lower than in experiments following direct excitation of ZnPc at, for instance, 680 nm.
:1 (see Fig. S-8†).20 Moreover, dilute solutions of PDI 1 (3.0 × 10−6 M)—in THF or benzonitrile (Fig. 2)—were titrated with variable amounts of ZnPc 5 (0–1.50 × 10−5 M) and probed by means of absorption and fluorescence spectroscopies. A 1:1 mixture in THF, for example, displays PDI centered (434, 527, and 568 nm) and ZnPc centered transitions (609, 632, 670, and 686 nm) in approximately a 1:3 ratio—identical with the simple superimposition of PDI 1 and ZnPc 5. Still, the presence of isosbestic points indicates the transformation of 1 (i.e., starting-point of the titration) into PDI/ZnPc (i.e., end-point of the titration). Less ambiguous were the corresponding excitation experiments following 526 nm excitation. Here, the PDI fluorescence, which evolves in the blue with a quantum yield of 1, decreased exponentially in the presence of variable concentrations of ZnPc 5. All throughout the titration, the PDI fluorescence is subject to a 60% quenching in THF, while in benzonitrile the overall quenching amounts to 40%. The exponential concentration/fluorescence relationship enabled the affinity constant, corresponding to the formation of PDI/ZnPc from PDI 1 and ZnPc 5, to be calculated. In THF and benzonitrile, the binding constants are 2 × 105 M−1 and 7 × 104 M−1, respectively. The fluorescence decrease is accompanied by a gradual blue shift of the main fluorescence band from 593 to 588 nm in THF and from 612 to 601 nm in benzonitrile. In the red part of the spectrum, namely around 700 nm, we see the characteristic ZnPc fluorescence emerging. The quantum yield of the latter—formed indirectly—is, however, with 0.063 (THF) and 0.074 (benzonitrile) markedly lower than in experiments following direct excitation of ZnPc at, for instance, 680 nm.
As a complement to the aforementioned steady-state fluorescence measurements we turned to time-resolved fluorescence spectroscopy. Here, the PDI fluorescence decay was probed at 610 nm—a wavelength where ZnPc 5 lacks any appreciable fluorescence—in the absence and presence of ZnPc 5. In the earlier scenario, that is, without ZnPc 5, the time–fluorescence profiles are best fitted by a single exponential decay function to yield lifetimes of 6.8 and 6.4 ns in THF and benzonitrile, respectively. These values are in excellent agreement with previously tested PDIs.7,12 Upon adding 1, 2, or even 5 equivalents of ZnPc 5, reasonable χ2 values were only obtained when using a biexponential fitting function. Common to all experiments is a slow and a fast decaying component. The slowly decaying constituent—with lifetimes around 6.7 ± 0.1 ns in THF and 6.1 ± 0.1 ns in benzonitrile—matches the intrinsic PDI fluorescence decay, while the fast part—with lifetimes of 3.0 ± 0.1 ns in THF as well as benzonitrile—reflects the deactivation with PDI/ZnPc.
Next, transient absorption spectroscopy (i.e., 150 fs laser pulses at 530 or 680 nm and 8 ns laser pulses at 355 or 532 nm) was employed to confirm the singlet excited state deactivations and, in addition, to characterize the nature of the photoproducts. Photoexciting PDI 1 at 530 nm (Fig. S-9†) leads to the population of the singlet excited state in the form of differential absorption changes, that include a broad transient bleach between 420 and 655 nm that is centered at 440 and 575 nm. In the red we see transient maxima at 700, 950, and 1030 nm (i.e., singlet–singlet transition). These singlet excited features convert via an inefficient intersystem crossing (i.e., with low quantum yields) to the energetically lower lying triplet excited features.8,9 From the intersystem crossing, we approximate a singlet lifetime of 6.0 ± 0.5 ns. Similarly, the metastable and faster decaying singlet–singlet features of ZnPc 5 (i.e., maxima at 500, 890, and 1275 nm; minima at 605, 660, and 650 nm)—formed upon 680 nm excitation—also give rise to an IC process—see Fig. S-10.†
When PDI/ZnPc was examined subsequent to laser excitation at either 530 or 680 nm, the same singlet excited state features observed in the case of PDI 1 or ZnPc 5 were immediately seen to develop—Fig. S-11.† This attests to the successful excitation of either PDI 1 or ZnPc 5. However, instead of the intersystem crossings, the PDI singlet excited state decays with lifetimes of about 3.0 ± 0.5 ns, a finding that is ascribed to an intraensemble energy transfer and this is in excellent agreement with the fluorescence measurements. Consistent with this conclusion, characteristics of the ZnPc singlet excited state were observed at the end of the decay, including transient minima at 605, 660, and 685 nm. The ZnPc singlet excited state in PDI/ZnPc, on the other hand, formed either indirectly—530 nm excitation experiments—or directly—680 nm excitation experiments, is stable. In fact, our time resolved experiments confirm that the ZnPc singlet excited state converts into the corresponding triplet excited state. Our experiments lacked, however, evidence for the involvement of a charge transfer reaction. In this context, we generated the one-electron oxidized ZnPc and the one-electron reduced PDI by means of pulse radiolysis with maxima at 520/840 and 700 nm, respectively.8,9 None of these features were seen.
In summary, we have applied the highly directional, triple hydrogen bonding of perylenediimides with ditopic melamines to design a supramolecular phthalocyanine–perylenediimide assembly. The relevance of energy transfer between non-covalently linked PDI and Pc units is demonstrated by our recent finding3 that high energy photons can be absorbed by a highly photoluminescent PDI chromophore undergoing Förster resonant energy transfer (FRET) to a Pc acting as an efficient DSSC-sensitizing dye.
Funding from MEC (CTQ2008-00418/BQU, CONSOLIDER-INGENIO 2010 CDS2007-00010 NANOCIENCIA MOLECULAR), COST Action D35, CAM (S-0505/PPQ/000225) and DFG (SFB 583) is acknowledged. M.S.R.-M. acknowledges MEC for a R&C research position.
Notes and references
- Molecular Mechanisms of Photosynthesis, ed. R. E. Blankenship, Blackwell Science, Oxford, UK, 2002 Search PubMed; J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 Search PubMed.
- G. de la Torre, C. G. Claessens and T. Torres, Chem. Commun., 2007, 2000–2015 RSC.
- B. E. Hardin, E. T. Hoke1, P. B. Armstrong, J.-H. Yum, P. Comte, T. Torres, J. M. J. Fréchet, M. K. Nazeeruddin, M. Grätzel and M. D. McGehee, Nat. Photonics, 2009, 3, 406–411 Search PubMed; B. C. O’Regan, I. López-Duarte, M. V. Martínez-Díaz, A. Forneli, J. Albero, A. Morandeira, E. Palomares, T. Torres and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 2906–2907 CrossRef CAS; J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martinez-Ferrero, E. Palomares, J. Ko, M. Graetzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358–8362 CrossRef CAS.
- Y. Rio, M. S. Rodríguez-Morgade and T. Torres, Org. Biomol. Chem., 2008, 6, 1877–1894 RSC and references therein.
- D. M. Guldi, J. Ramey, M. V. Martínez-Díaz, A. de la Escosura, T. Torres, T. Da Ros and M. Prato, Chem. Commun., 2002, 2774–2775 RSC; D. M. Guldi, I. Zilbermann, A. Gouloumis, P. Vázquez and T. Torres, J. Phys. Chem. B, 2004, 108, 18485–18494 CrossRef CAS.
- X. Li, L. E. Sinks, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 10810–10811 CrossRef CAS.
- M. S. Rodríguez-Morgade, T. Torres, C. Atienza Castellanos and D. M. Guldi, J. Am. Chem. Soc., 2006, 128, 15145–15154 CrossRef CAS.
- A. J. Jimenez, F. Spänig, M. S. Rodríguez-Morgade, K. Ohkubo, S. Fukuzumi, D. M. Guldi and T. Torres, Org. Lett., 2007, 9, 2481–2484 CrossRef CAS.
- S. Fukuzumi, K. Ohkubo, J. Ortiz, A. M. Gutierrez, F. Fernandez-Lazaro and A. Sastre-Santos, Chem. Commun., 2005, 3814–3816 RSC.
- J. E. Norton and J.-L. Bredas, J. Chem. Phys., 2008, 128, 034701-1–034701-7; J. Tant, Y. H. Geerts, M. Lehmann, V. De Cupere, G. Zucchi, B. W. Laursen, T. Bjornholm, V. Lemaur, V. Marcq, A. Burquel, E. Hennebicq, F. Gardebien, P. Viville, D. Beljonne, R. Lazzaroni and J. Cornil, J. Phys. Chem. B, 2005, 109, 20315–20323 CrossRef CAS.
- S. Campidelli, B. Ballesteros, A. Filoramo, D. Diaz Diaz, G. de la Torre, T. Torres, G. M. A. Rahman, C. Ehli, D. Kiessling, F. Werner, V. Sgobba, D. M. Guldi, C. Cioffi, M. Prato and J.-P. Bourgoin, J. Am. Chem. Soc., 2008, 130, 11503–11509 CrossRef CAS; M. S. Rodríguez-Morgade, M. E. Plonska-Brzezinska, A. J. Athans, E. Carbonell, G. de Miguel, D. M. Guldi, L. Echegoyen and T. Torres, J. Am. Chem. Soc., 2009, 131, 10484–10496 CrossRef CAS and references therein.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- Y. Chen, Y. Lin, M. E. EI-Khouly, X. Zhuang, Y. Araki, O. Ito and W. Zhang, J. Phys. Chem. C, 2007, 111, 16096–16099 CrossRef CAS; F. J. Cespedes-Guirao, K. Ohkubo, S. Fukuzumi, A. Sastre-Santos and F. Fernandez-Lazaro, J. Org. Chem., 2009, 74, 5871–5880 CrossRef CAS.
- L. Sánchez, N. Martín and D. Guldi, Angew. Chem., Int. Ed., 2005, 44, 5374–5382 CrossRef CAS; J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS.
- G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. N. Chin, M. Mammen and D. M. Gordon, Acc. Chem. Res., 1995, 28, 37–44 CrossRef CAS.
- C. Thalacker and F. Würthner, Adv. Funct. Mater., 2002, 12, 209–218 CrossRef CAS; L. E. Sinks, B. Rybtchinski, M. Iimura, B. A. Jones, A. J. Goshe, X. B. Zuo, D. M. Tiede, X. Y. Li and M. R. Wasielewski, Chem. Mater., 2005, 17, 6295–6303 CrossRef CAS; J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS; F. J. M. Hoeben, J. Zhang, C. C. Lee, M. J. Pouderoijen, M. Wolffs, F. Würthner, A. P. H. J. Schenning, E. W. Meijer and S. De Feyter, Chem.–Eur. J., 2008, 14, 8579–8589 CrossRef.
- F. Würthner, C. Thalacker and A. Sautter, Adv. Mater., 1999, 11, 754–758 CrossRef CAS.
- E. M. Maya, P. Vázquez and T. Torres, Chem.–Eur. J., 1999, 5, 2004–2013 CrossRef CAS.
- G. Ricciardi, A. Rosa and E. J. Baerends, J. Phys. Chem. A, 2001, 105, 5242–5254 CrossRef CAS.
- J. L. Sessler, J. Jayawickramarajah, A. Gouloumis, G. D. Pantos, T. Torres and D. M. Guldi, Tetrahedron, 2006, 62, 2123–2131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for the preparation of unreported compounds, as well as NMR, IR, UV/Vis, MS and transient absorption spectra. See DOI: 10.1039/b921363e |
| This journal is © The Royal Society of Chemistry 2010 |