Study on microscopic homogeneity of polymeric candidate reference materials BAM H001–BAM H010 by means of synchrotron µ-XRF and LA-ICP-MS
Christoph
Simons
a,
Christian
Mans
*ad,
Stephanie
Hanning
a,
Anton
Janßen
a,
Martin
Radtke
b,
Uwe
Reinholz
b,
Markus
Ostermann
b,
Matthias
Michaelis
b,
Julia
Wienold
b,
Dorothea
Alber
c and
Martin
Kreyenschmidt
a
aUniversity of Applied Sciences Münster, Department of Chemical Engineering, Advanced Analytical Chemistry, Stegerwaldstr. 39, 48565, Steinfurt, Germany. E-mail: c.mans@fh-muenster.de; Fax: +49 (0) 25 51/9-62-429; Tel: +49 (0) 25 51/9-62-581
bFederal Institute for Materials Research and Testing (BAM), Richard-Willstätter-Straße 11, 12489, Berlin, Germany
cHelmholtz-Zentrum Berlin, Department SF6, Glienicker Straße 100, 14109, Berlin, Germany
dTechnical University Bergakademie Freiberg, Faculty for Chemistry and Physics, Institute for Analytical Chemistry, Leipziger Straße 29, 09599, Freiberg, Germany
First published on 19th November 2009
Abstract
In this study the microscopic homogeneity of the newly developed plastic reference materials BAM H001–BAM H010 was investigated. The materials consist of an acrylonitryle-butadien-styrene terpolymer, doped with different amounts of the elements Br, Cd, Cr, Hg and Pb. For the quantitative determination of the degree of homogeneity, a procedure proposed by Kempenaers et al. (Fresenius J. Anal. Chem., 2001, 369, 733–737) was used. On every sample an extensive number of different points were analyzed and standard deviation for every element mentioned above was used to calculate a minimal sampling mass that is necessary to reach a certain level of uncertainty caused by inhomogeneity (mmin,5%). The experiments were taken out with synchrotron µ-XRF (SR µ-XRF) at BESSYII in Berlin and by laser ablation inductively coupled plasma mass spectroscopy (LA-ICP-MS). The calculated values for mmin,5% of both techniques showed comparable results for all elements. It could be shown that the materials are suitable for calibration of micro analytic techniques if at least 64 µg are used.
Introduction
Polymers are essential materials in our modern society. Due to their use in numerous fields of applications, polymers are subjected to a number of restrictions and regulations regarding the presence and concentration of various additives and fillers. The need for high sample throughput in product control and cost effectiveness demands fast and reliable analytical methods for a number of elements.Methods of direct solid sampling such as LA-ICP-MS or XRF are well established beside the classical employed methods for elemental analysis such as ICP-MS following digestion.1 Unfortunately measurements with the aid of solid sampling methods are often influenced by the matrix of the samples.2
Therefore, calibration materials which match the sample matrix as close as possible are needed. Furthermore these materials need to be well characterized regarding the analyte content, homogeneity and stability. This also counts for materials used for the validation of analytical procedures. For these materials a certification of the properties stated above is necessary.
A new set of candidate reference materials was produced to satisfy the demands on suitable reference materials to determine element traces in polymers.
The set consists of 10 materials based on acrylonitrile-butadiene-styrol terpolymer, doped with different contents of the elements Br, Cd, Cr, Hg and Pb ranging from 2 to 1500 mg/kg. The production procedures and the preliminary characterization of the elemental content of these materials were already described in the literature.3–5 The mass fractions of each material determined by NAA and ICP-MS following digestion are listed in Table 1.
| Sample | Br | Cd | Cr | Hg | Pb |
|---|---|---|---|---|---|
| a Uncertainty of NAA results is about 7%. | |||||
| BAM H001 | <0.1 | <2 | <1 | <0.15 | <0.5 |
| BAM H002 | 1419 | 19 | 461 | 4 | 93 ± 0.7 |
| BAM H003 | 98 | 10 | 977 | 11 | 495 ± 6.3 |
| BAM H004 | 50 | 7 | 101 | 17 | 1434 ± 8.0 |
| BAM H005 | 938 | 183 | 47 | 33 | 16 ± 0.4 |
| BAM H006 | 25 | 100 | 16 | 63 | 954 ± 13.2 |
| BAM H007 | 6 | 26 | 1483 | 396 | 60 ± 1.3 |
| BAM H008 | 466 | <7 | 7 | 878 | 24 ± 0.7 |
| BAM H009 | 19 | 49 | 27 | 1314 | 5 ± 0.1 |
| BAM H010 | 234 | 86 | 510 | 421 | 482 ± 5.9 |
A total mass of 150 kg of each material was produced as granulates. A part of these materials was then used to produce solid bodies with a diameter of 40 mm and different thicknesses of 1 mm, 2 mm, and 6 mm (500 units per batch and thickness).
In this current study, the microscopic homogeneity of this set of candidate reference materials was investigated by means of LA-ICP-MS and SR µ-XRF prior to the certification process.
Further on, the homogeneity of a material will be expressed by the minimal necessary sample mass, which needs to be analyzed to reach a given level of uncertainty caused by inhomogeneity of the sample e.g. 5% (mmin5%).
For the determination of this value, analytical methods with high lateral resolution like LA-ICP-MS or µ-XRF have to be applied. The uncertainty of a number of measurements with small sample masses is determined and the correlation of the uncertainty and the sample mass is explored. This stotal(m)-graph can be described by a power function
| stotal(m) = am−b + c | (1) |
The three coefficients a, b and c can be derived from a fitting procedure. mmin,5% can then be estimated from setting stotal(m) = 5%.
A detailed description of the concept used for examination of microscopic homogeneity including the definition of all coefficients was already published by Kempenaers et al.6–10
Experimental
SR-µ-XRF
Micro homogeneity was assessed using µ-XRF with synchrotron radiation (SR µ-XRF) at the µSpot Line and BAM-Line at BESSYII in Berlin.11A sketch of the experimental setup is shown in Fig. 1, the used parameters are listed in Table 2.
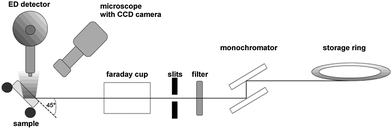 | ||
| Fig. 1 Setup of the µ-XRF measurements at BESSYII. | ||
| Cd | Cr | Hg | Pb | Br | |
|---|---|---|---|---|---|
| For samples H003, H004, H007, H008, H010 | |||||
| Setup | BAM-Line | µSpot | µSpot | µSpot | µSpot |
| Excitation in keV | 40 | 9.5 | 20 | 20 | 20 |
| Spot size in µm | 60 × 125 | 70 × 95 | 70 × 95 | 70 × 95 | 70 × 95 |
| Analyzed mass per spot in µg | 9 | 8 | 8 | 8 | 8 |
| Measured line | Kα | Kα | Lα | Lα | Kβ |
| For samples H002, H005, H006, H009 | |||||
| Setup | µSpot | µSpot | µSpot | µSpot | µSpot |
| Excitation in keV | 20 | 20 | 20 | 20 | 20 |
| Spot size in µm (diameter) | 100 | 100 | 100 | 100 | 100 |
| Analyzed mass per spot in µg | 4.2 | 4.2 | 4.2 | 4.2 | 4.2 |
| Measured line | Lα | Kα | Lα | Lα | Kβ |
The measurements were performed in the standard 45°/45° geometry to minimize scattering from the samples. A HPGe detector was deployed and measurement time was optimized to reach approximately 1 × 104 counts for every element. Due to limited operation time, this could not be achieved for every element in each sample.
The X-ray spectral fittings were performed using the QXAS software.12 The obtained peak areas were normalized to detector dead time and excitation intensity.
A raster of 21 × 21 points with a distance of 200 µm was measured on each sample with a thickness of 1 mm. The methods standard deviation for each setup was examined by measuring 441 times on an individual spot on each sample.
The analyzed mass per single spot was calculated from the previously determined spot size, sample thickness and the density of the ABS-Material (1.17 g/cm3).
The normal distribution was proofed for the acquired data, using the Shapiro-Wilk test with a confidence interval of 95%. Higher sampling masses were achieved by summing up data pair wise.
The standard deviation of each data set was calculated and corrected with the previously determined smeth gaining the standard deviation caused by the inhomogeneity of the material. Formula 1 was then fitted to the data, and the parameters a, b and c were determined. Once the formula was known for each material and element, mmin,5% was calculated (Table 3).
| Cr | Cd | Br | Pb | Hg | |
|---|---|---|---|---|---|
| a count rate too low. b mmin,5% < sampling mass. c smeth > stotal. | |||||
| BAM H002 | 8.1 | <4.2a3 | <4.2c | <4.2b | |
| BAM H003 | 21 | <9b | <8b | <8b | <8b |
| BAM H004 | 64 | <8b | <8b | ||
| BAM H005 | <4.2b | <4.2b | |||
| BAM H006 | <4.2b | <4.2c | |||
| BAM H007 | <8a2 | <9b | <8b | <8b | |
| BAM H008 | <8 | <8b | <8b | ||
| BAM H009 | <4.2b | ||||
| BAM H010 | 19 | <9b | <8b | <8b | 31 |
| BAM H010 by LA-ICP | <1 | <1 | <1 | <1 | 28 |
| BCR 680 Literature6 | 29 | 3 | 1 | 9 | 3 |
LA-ICP-MS
Additionally to the XRF measurements the microscopic homogeneity of the BAM H010 sample was determined via LA-ICP-MS as an alternative technique.A 213 nm laser (CETAC) in combination with an ICP-MS (Elan DRC II, Perkin Elmer) was applied for the measurements of a sample with a thickness of 1 mm.
25 measurements were carried out at different locations in a raster of 5 × 5 points with a distance of 1 mm between the single spots. The spot size was 200 µm in diameter. Measurements were carried out applying the parameters listed in Table 4. The ablated mass per laser shot was 16.8 ng as determined in an earlier study.13
| Laser Parameter | |
| Laser Voltage | 715 V (100%) |
| Repetition Rate | 20 Hz |
| Ablations per Spot | 1000 |
| Ablation Gas | Helium |
| Sweep Gas | Argon |
| Sweep Gas flow | 1 L/min |
| Ablation Modus | Spot Ablation |
| Shutter time | 10 s |
| Time for Gas Blank | 10 s |
| Measurement delay ICP | 20 s |
| Measurement time for each spot | 40 s |
| ICP-MS Parameter | |
| Auxilary gas flow | 1 L/min |
| Plasma Gas flow | 15 L/min |
| ICP RF-Power | 1250 W |
| Isotope | 79Br, 13C, 114Cd, 53Cr, 202Hg, 206Pb |
| Dwell-time | 50 ms |
| Measurement cycles per point | 100 |
The data for all elements was divided by the measured intensity of the 13C isotope, which was used as an internal standard.13 In addition Hg, Cr and Cd signals were corrected for drifts over time by a polynomial fit as proposed by Kempenaers et al.7 The determined fraction indices resulted in volumes between 0.96 and 1.02.
For every measurement point, a time (and depth) resolved signal was acquired. To gain data for different sampling masses, average intensities of this signals were calculated from all data points after 3, 5, 10, 20, 30 and 40 seconds of measurement time. This leads to sampling portions of 1.0 µg, 1.7 µg, 3.4 µg, 6.7 µg, 10.01 µg, 13.4 µg respectively 16.8 µg.
The determination of mmin,5% was carried out as described above and in Ref. 7.
Results and discussion
The microscopic homogeneities of the analytes in the candidate reference materials BAM H001–BAM H010 were assessed with SR µ-XRF and LA-ICP-MS. A value for mmin,5% was calculated for each material and the results are summarized in Table 3.The determination of mmin,5% is limited by the smallest mass, which can be sampled by the method applied.
For LA-ICP-MS this mass correlates with the diameter of the crater, the number of shots, which is carried out during a data acquisition cycle and the ablation rate. The ablation rate was previously determined for the sample matrix ABS and the employed LA setup.13 The lowest mass that could be resolved during the experiments was 1 µg.
In SR µ-XRF the smallest mass of sample, which can be resolved during the study was determined by the diameter of the attenuating X-ray beam and the depth of penetration of the X-ray beam. For polymer matrices with low mass attenuation coefficients the probed depth can be assumed to be equal to the samples thickness.
The minimal sampled mass differs between the SR µ-XRF experiments as different setups were applied. According to the different spot sizes, the lowest resolvable masses range from 4.2 to 9 µg.
Due to the given measurement time at BESSYII a sufficient number of counts could not be achieved for every element in each sample. Whenever, the absolute counts were <2000, the elements were not taken into account for this study and were therefore, indicated as ‘count rate too low’.
For Br and Pb the calculated values for mmin,5% were always lower than the smallest probed mass (<9 µg, <8 µg and <4.2 µm respectively) in the SR µ-XRF experiments. This is in good correlation with results gained by the LA-ICP-MS for BAM H010 where mmin,5% < 1 µg was determined.
The very homogeneous distribution of Br and Pb was expected due to the good solubility of the used species. Pb was worked into the ABS as stearate and Br as the organic compound decabromodiphenylic ether. Both were supposed to show a good solubility in the polymeric melt. Previous studies have already indicated that organic species of the analytes should be preferred for the production of polymeric calibration materials whenever highly homogeneous distributions are required, while inorganic species often need further treatment to obtain sufficiently small particles.13
The determined values of mmin,5% gained for Hg were satisfying as well. mmin,5% was determined to be <8 µg for all measured samples with the exception of BAM H010 where a value for mmin,5% of 31 µg (SR µ-XRF) respectively 28 µg (LA-ICP-MS) was determined.
In comparison to Br, Cr, Pb and Hg the excitation of Cd for SR µ-XRF measurements requires a higher energy for the Kα line. For this reason, the BAM-line was used to measure Cd in BAM H003, BAM H007 and BAM H010 with an exciting energy of 40 keV in a separate experiment. The results for mmin,5% were very satisfying for this element. For all three samples where mmin,5% was examined, the determined values were smaller than the smallest mass probed. These results were in good correlation to the results determined with LA-ICP-MS.
Cr-Kα possesses the lowest fluorescence energy compared to the other elements measured in this study. For this reason Cr had to be measured in a separate experiment using µSpot-Line applying an optimized excitation energy of 9.5 keV. The resulting mmin,5% varied in a range from <8 to 64 µg.
Cr showed the highest heterogeneity of the investigated analytes. Data derived from XRF measurements of Cr showed a highest fluctuation compared to the measurements of all other elements investigated.
In Fig. 2 the corrected intensities of 441 SR µ-XRF measurements for Cr are compared to the corrected intensities obtained for Cd in the same sample. The intensities gained for Cd showed less fluctuations. In contrast, the signals for Cr measured at different locations of the sample showed numerous spikes.
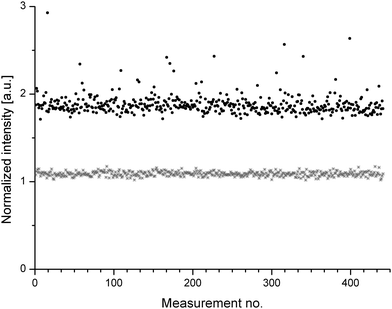 | ||
| Fig. 2 Data plot from 441 SR y-XRF experiments for the elements Cr (black) and Cd (grey) in sample BAM H010. Data was corrected via scattering and for Cd an offset of 0.5 units was introduced for better visualisation. | ||
Tests of this data for normal distribution by a Shapiro-Wilk test indicated that the data is not distributed randomly. This indicates the presence of centers in the samples with high Cr content, which might be caused by the presence of Cr2O3 nuggets. Beside the observation of the spikes in intensity, also the other data points measured for Cr showed a higher fluctuation. These two effects have resulted in higher values of mmin,5% for Cr. Nevertheless, Kempenaers et al. showed in a former study, that the values of mmin,5% are not significantly affected by the presence of small nuggets.6
Measurements with LA-ICP-MS on sample BAM H010 revealed smaller values of mmin,5% for Cr. In contrast to the SR µ-XRF measurements no spikes were observed.
Compared to SR µ-XRF, LA-ICP-MS samples a much smaller volume. Therefore, the probability of this method to miss nuggets in a sample is much higher. This indicates that measurements by LA-ICP-MS can suppress the presence of nuggets in samples, especially, if the number and sizes of these nuggets are small.
The size of the nuggets was estimated with the aid of a scanning electron microscope in combination with an energy dispersive X-ray detector (SEM-EDX). A SEM-EDX picture of sample BAM H010 is shown in Fig. 3. Particles with diameters of app. 1 µm, visible as white spots, are distributed over the observed sample area. EDX measurements of theses spectra revealed the presence of high Cr concentration in these particles. No nuggets were found for any of the other investigated elements. This implies, that only Cr was not entirely homogeneously distributed in the polymeric matrix, while all other elements were distributed to an extent that they could not be observed further under the applied SEM resolution.
 | ||
| Fig. 3 SEM picture of the sample BAM H010 with an EDX spectrum of one prominent particle (see arrow). | ||
Conclusion
The distributions of the elements Br, Cd, Cr, Hg and Pb in the new set of polymeric candidate reference materials were investigated with the aid of SR µ-XRF and LA-ICP-MS. The elements can be estimated to be sufficiently homogeneously distributed, if at least 64 µg of the sample are probed.For a typical XRF measurement this homogeneity is more than sufficient, as usually several grams are analyzed. For a typical LA-ICP-MS measurement with an approximately sampling rate of 20 ng per shot, 3500 shots are necessary.
The values determined for mmin,5%via SR µ-XRF and LA-ICP for sample BAM H010 showed a good comparability for all measured elements except for Cr.
It could be shown that the produced materials contain nuggets of Cr2O3 in a size up to 1 µm.
Furthermore, it could be shown, that organic and metal organic species show a better distribution in polymeric samples as inorganic species. Inorganic species should be used with the smallest possible particle size, when a very high homogeneity is required. This observation was also made in a former study.13
Comparing the examined values for mmin,5% of the newly developed samples to BCR 680 one can conclude that the achieved homogeneity is in the same order of magnitude.
All in all it can be concluded that the produced materials show a sufficient microscopic homogeneity, and that they can be used for calibration of several analytic techniques.
Acknowledgements
The authors wish to thank Mr. Thorsten Grund (Institute for Planetology of University Münster) for carrying out the SEM measurements.References
- J. S. Becker, Spectrochim. Acta, Part B, 2002, 57, 1805–1820 CrossRef.
- R. E. Russo, X. Mao, H. Liu, J. Gonzalez and S. S. Mao, Talanta, 2002, 57, 425–451 CrossRef CAS.
- Application: WO, 2006–DE1970 2007056977, 2007.
- C. Mans, C. Simons, S. Hanning, A. Janssen, D. Alber, M. Radtke, U. Reinholz, A. Buehler and M. Kreyenschmidt, X-Ray Spectrom., 2009, 38, 52–57 CrossRef CAS.
- C. Mans, S. Hanning, C. Simons, A. Wegner, A. Janssen and M. Kreyenschmidt, Spectrochim. Acta, Part B, 2007, 62, 116–122 CrossRef.
- L. Kempenaers, C. De Koster, W. Van Borm and K. Janssens, Fresenius J. Anal. Chem., 2001, 369, 733–737 CrossRef CAS.
- L. Kempenaers, N. H. Bings, T. E. Jeffries, B. Vekemans and K. Janssens, J. Anal. At. Spectrom., 2001, 16, 1006–1011 RSC.
- L. Kempenaers, L. Vincze and K. Janssens, Spectrochim. Acta, Part B, 2000, 55, 651–669 CrossRef.
- L. Kempenaers, K. Janssens, K. P. Jochum, L. Vincze, B. Vekemans, A. Somogyi, M. Drakopoulos and F. Adams, J. Anal. At. Spectrom., 2003, 18, 350–357 RSC.
- L. Kempenaers, K. Janssens, L. Vincze, B. Vekemans, A. Somogyi, M. Drakopoulos, A. Simionovici and F. Adams, Anal. Chem., 2002, 74, 5017–5026 CrossRef CAS.
- W. Gorner, M. P. Hentschel, B. R. Muller, H. Riesemeier, M. Krumrey, G. Ulm, W. Diete, U. Klein and R. Frahm, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468, 703–706 CrossRef CAS.
- Quantitative X-ray System 2.0 ( 2005), International Atomic Energy Agency, IAEA, Vienna, Austria, http://www.iaea.org Search PubMed.
- C. Simons, S. Hanning, A. Wegner, C. Mans, A. Janßen and M. Kreyenschmidt, J. Anal. At. Spectrom., 2008, 23, 1038–1041 RSC.
| This journal is © The Royal Society of Chemistry 2010 |