Indirect determination of trace amounts of lithium via complex with iron by X-ray fluorescence spectrometry
Beata
Zawisza
*
Institute of Chemistry, Silesian University, ul. Szkolna 9, 40-006, Katowice, Poland. E-mail: beata.zawisza@us.edu.pl; Fax: +48 32 2599978
First published on 8th October 2009
Abstract
The extremely low fluorescent yield and long-wavelength characteristic radiation are the major limitations for direct XRF determination of the elements of low atomic numbers. In the case of lithium, the determination of this element by X-ray fluorescence spectrometry (XRF) is not possible. Thus, an indirect method for the determination of lithium by X-ray fluorescence spectrometry was proposed. The method is based on the determination of lithium through iron in the potassium lithium periodatoferrate(III) complex. The complex was collected on the membrane filter and the Fe Kα line was measured by XRF. The optimum conditions for LiKFeIO6 complex formation and XRF measurement were studied. A good ratio of lithium to iron (1 to 8) and a sensitive Fe Kα line give the possibility to determine a low lithium amount. The proposed method allows the determination of 20 µg of lithium collected on a filter. The validity of the proposed method was verified with certified lithium ores. The agreement between XRF analysis and certified values is satisfactory and indicates the usefulness of the carried out method.
Introduction
The determination of lithium in rock and minerals is of interest in geochemical exploration. Lithium is used as trace metal for the identification of the ratio of natural/antropogenic metal in sedimentary mineral deposits.1 Lithium as the minor sea water cationic component is also frequently determined.2 The precise determination of the isotopic composition of a natural material containing Li and heavy metals is very important for the environment,3 especially as a guide to hydrothermal alteration, progressive metamorphism and hydrothermal water-sediment-rock interaction. Lithium is frequently determined in different kinds of water, i.e. lake water,4 sub-surface water, ground water and sea water or natural brines.5,6 The therapeutic effects of lithium salts are also well known, for instance, in the treatment of manic depressive patients.7,8The main used techniques for the determination of lithium are flame atomic absorption spectrometry (AAS),9 thermal ionization mass spectrometry (TIMS),3 inductively coupled plasma atomic emission spectrometry (ICP-AES),10 flame emission spectrometry (FAES),11 differential pulse anodic stripping voltammetry (DPASV),12 chromatography,4 capillary electrophoresis (CE),2 flow injection analysis (FIA)13 and spectrophotometry.14 Nevertheless, the use of most of them is connected with some unwelcome effects and difficulties for the precise determination of lithium. Thus, the influence of the ions Na+, K+, Ca2+, Mg2+ on the determination of lithium is required for AAS. A depressive effect on the lithium determination rises parallel to the increase in the content of the aforementioned interfering ions. The same elements, especially sodium and potassium, can interfere with the results obtained by ICP-AES due to a decrease of lithium ionization, scattering and overlapping spectral emissions. Thus, taking the interferences into consideration, it is justified to use the standard addition method for determining lithium in biological fluids. Sodium is also a problem in the determination of lithium e.g. in sea water by isotopic analysis, which require a high purity of Li. Separation of Li can be carried out by cation-exchange chromatography, but the major difficulty is residual sodium, which overlaps with Li. Thus, the samples with Na need a much higher ionization filament current to form a stable Li+ ion beam intensity. Positive response to interference on the electrode can be also observed in voltammetric methods. Moreover, these techniques usually involve a selective preconcentration process of trace amounts of lithium ions.
In this work, I would like to propose an indirect method for the determination of lithium by X-ray fluorescence spectrometry (XRF). The accurate, direct determination of this element by X-ray fluorescence spectrometry is impossible because of the emission of very long-wavelength radiation and extremely low fluorescent yield of this element. Thus, lithium is undetectable by XRF spectrometry. An indirect method was described for the determination of lithium after precipitating as arsenate or phosphate.15 The method proposed in this paper is based on the determination of the lithium by precipitating it in the potassium lithium periodatoferrate(III) complex16,17 and measuring the iron by X-ray fluorescence spectrometry. The lithium in this method is determined indirectly via measurement of the sensitive Fe Kα line. The worked out procedure allows for the determination with high precision and accuracy of minor amounts of lithium. The developed method was verified with certified lithium ores.
Experimental
Materials
All reagents are from Merck (Germany). Milli-Q grade purity water was used.The precipitating agent: 2.3 g of KIO4 was dissolved in 50 mL of 0.5 M solution of KOH, next 8 mL of 0.05 M FeCl3 solution in 2 M HCl was added; the mixture was filled up by 2 M KOH to 100 mL.
Standard Reference Material of Lithium Ores 181 and 182 from the National Institute of Standards and Technology.
Membrane filters (1.2 µm pore size, White RAWP, 25 mm diameter) from Millipore (USA).
Apparatus
XRF measurements were performed using a wavelength-dispersive sequential X-ray spectrometer with a silver target X-ray tube which was operated at 50 kV and 40 mA. The incidence and take-off angle were ψ1 = 60° and ψ2 = 40°, respectively. LiF (200) analyzing crystal, flow-proportional detector and fine collimator were used. The counting time was 20 s. Each sample was rotated while measurements were taken to avoid the effect of any inhomogeneity of the mass per unit area. The measurements were performed in vacuum. The net intensities were determined for each sample by the measurement of fluorescent radiation of the element (peak) in the analyzed samples and the blank sample.The precipitates were filtered off using a filtration assembly (25 mm, Millipore).
Synthetic sample preparation
1.0 mL of 50 µg mL−1 solution of lithium was transferred into 25 mL beakers. Then 1 mL of 1 M potassium hydroxide solution was added. The solution was heated to boiling. Then 3 mL of potassium periodate/ferric chloride solution heated to 90 °C was added. The obtained solution was heated to about 90 °C for 5 minutes. After cooling down, the precipitate was filtered off in the filtration assembly with Millipore membrane filters and then it was rinsed with 4 × 2 mL portions of 1 M potassium hydroxide solution. The loaded filter was mounted on a plastic ring by means of a two-sided adhesive tape and dried using an IR heater at 60 °C. After drying out, a few drops of a 0.5% solution of polystyrene in carbon tetrachloride were dropped into the sample to protect it. After drying, the sample is durable and can be analyzed many times by XRF spectrometry.The blank sample preparation was identical to this described above except that water was added instead of the lithium solution.
Effect of excess of potassium periodate/ferric chloride solution
0.5, 1.0, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0 and 7.0 mL of potassium periodate/ferric chloride solution were added into nine beakers with 50 µg of lithium. For each sample, a blank sample was prepared separately with the right amount of potassium periodate/ferric chloride. The procedure of the precipitation of potassium lithium periodatoferrate(III) complex is described in detail in the Synthetic Sample Preparation section.Effect of sodium, rubidium, aluminium and alkaline-earth metals on the potassium lithium periodatoferrate(III)
The elements were chosen due to their common occurrence in the various geological materials.A 1.0 mL aliquot of a 50 µg mL−1 solution of lithium was transferred into twenty three beakers. Then different amounts of solutions of sodium, rubidium, magnesium, calcium, strontium, barium and aluminium were added separately, i.e.
a) 50, 500, 5000, 10000 and 50000 µg of sodium
b) 500, 1000 and 5000 µg of rubidium
c) 250, 1250 and 2500 µg of magnesium, calcium, strontium, barium
d) 2500, 5000 and 12500 µg of aluminium.
The subsequent steps of the precipitation procedure and the others reagents are described in the Synthetic Sample Preparation section.
Effect of some elements on the potassium lithium periodatoferrate(III)
The elements were chosen for their stereochemistry and ionic radius, which might permit their inclusion in the periodatoferrate(III) complex. A 1.0 mL aliquot of a 50 µg mL−1 solution of lithium was transferred into fourteen beakers. In the sample preparation process, different amounts of 1.0 mg mL−1 of Ni, Ga, Cr, Bi, Co and Mn solutions were added separately, i.e. 250, 500 and 5000 µg. The subsequent steps of the precipitation procedure and the others reagents are identical to those described in the Synthetic Sample Preparation section.Preparation of calibration standards
20, 30, 40, 50, 60 and 70 µg of lithium solution were transferred into six beakers. The standards preparation was identical to the synthetic sample preparation.Standard reference material preparation
1.5 mg of standard reference material 181 and 182 (lithium ores) were digested in 10 mL (by 1 mL portions) of concentrated hydrofluoric acid. After evaporation to dryness of the samples, 10 mL (by 2 mL portions) of concentrated hydrochloric acid were added. After digestion of the minerals, the obtained solutions were transferred into two 25 mL beakers, and lithium was precipitated according to the procedure described in the Synthetic Sample Preparation section.Determination of precipitation yield of lithium
The recovery of lithium after the precipitation was checked on the basis of both, the Standard Reference Materials of Lithium Ores and synthetic standards (30 µg of Li). The details of the sample preparation are described in the Synthetic Sample Preparation section and Standard Reference Material Preparation section. After precipitation of the potassium lithium periodatoferrate(III) complex and filtration of the precipitates, lithium was determined in the filtrates by ICP-OES.Repeatability
Seven samples of each synthetic standard with 30 µg of lithium as well as seven samples of Standard Reference Material of Lithium Ore 182 (digestion of ca. 1.5 mg of the lithium ore in the concentrated hydrofluoric acid and concentrated hydrochloric acid) were prepared. The details of the sample preparation are described in the Synthetic Sample Preparation section and Standard Reference Material Preparation section, respectively.Results and discussion
The carried out method is based on using the following reaction: Li+ + IO4− + FeCl3 + 4KOH → KLiFeIO6↓ + 3KCl + 2H2O and the evaluation of the lithium in the formed potassium lithium periodatoferrate(III) complex by determination of lithium via measurement of the Fe Kα line by XRF. The ratio of lithium to iron (m/m) in the above mentioned complex is very favourable (1 to 8), which makes the method sensitive, suitable for the determination of trace amounts of lithium and suitable for obtaining very good detection limits. Not only the profitable ratio of lithium to iron in the stoichiometric potassium lithium periodatoferrate(III) complex but also the sensitive Fe Kα line are of great importance in the precise indirect determination of lithium by XRF spectrometry.The optimization of the precipitation process required the specification of the temperature of reagents, the time of ageing of the precipitate and the excess of precipitating agent (potassium periodate/ferric chloride solution). Both, the temperature of the added reagents and the time of heating of the mixtures (5–15 min) do not influence the XRF measurement of Fe Kα and, in consequence, the indirect determination of lithium in LiKFe(IO6). Nevertheless, a fawn-yellow solid appeared only when the mixture was heated. Heating at least for 5 minutes was enough to complete the precipitation of the potassium lithium periodatoferrate(III) complex. The hot precipitant was added and then the obtained solution was heated to about 90 °C for 5 minutes. The complete and quantitative precipitation of lithium required examination of amount of added precipitating agent, especially the ferric chloride solution. Iron as 4·10−3 M FeCl3 (in the mixture with 0.1 M KIO4) was added in excess from 0.8 to 31.5 (m/m) in relation to lithium in order to evaluate the effect of this parameter on the precipitation of the complex. Fig. 1 shows the results of this research. The complete precipitation of lithium was obtained when a 11.2 excess of Fe3+ was used, what in this considered case (50 µg of Li+) means 2.5 mL of KIO4 + FeCl3 solution (47.75 mg of IO4− and 0.560 mg of Fe3+). The obtained amount of precipitant is also sufficient for quantitative determination of 70 µg of Li+. In the carried out method, a bit larger excess of precipitating agent was used (13.44 excess of Fe3+) i.e. 3 mL, which means 57.3 mg of IO4− and 0.672 mg of Fe3+.
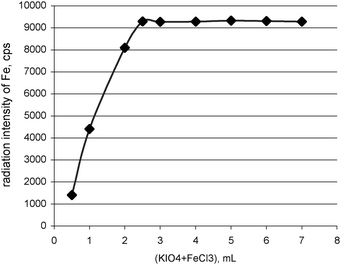 | ||
| Fig. 1 Effect of excess of potassium periodate/ferric chloride solution on the recovery of lithium. | ||
The influence of some elements on LiKFe(IO6) was studied and the results of those studies are presented in figure 2. The elements were chosen due to their common occurrence and the possibility of the formation of a similar complex to the potassium lithium periodatoferrate(III) complex. Thus, the common occurrence of sodium, rubidium, aluminium, magnesium, calcium, strontium, barium in the different geological materials and giving in alkaline solution insoluble hydroxides or similar complexes were the reasons why these elements were chosen. For example, the LiMFeIO6 (M = K, Rb or Cs) complexes can be formed in certain conditions.18 The conducted research indicates that in the case of the proposed method, rubidium does not precipitate and does not influence the radiation intensity of Fe Kα. A trace amount of sodium appears in the precipitate but in 100 times excess in relation to lithium does not disturb the determination of lithium via Fe Kα measurement. The large amount of sodium (>5 mg) affects indirect determination of lithium for the sake of the high blank sample. The co-precipitation of some compounds of sodium in the blank samples can be the cause of the adsorption of excessive amount of iron on the precipitate and, thereby, it makes the indirect determination of lithium by iron difficult or even impossible. Carried out experiments also indicate that aluminium and alkaline metals except for magnesium precipitate along with the analyzed complex. Calcium, strontium, barium and aluminium give in alkaline solution hydroxides, but this fact does not influence the radiation intensity of Fe if the amount of Ca, Sr and Ba does not excess in relation to Li 25:1 and 100:1 in the case of Al. The larger amount of these elements causes the adsorption of excessive amounts of iron on the surface of the samples, especially blank samples. In the case of 2.5 mg Mg (50:1 Mg:Li), only an insignificant amount of magnesium was found in the analyzed complex.
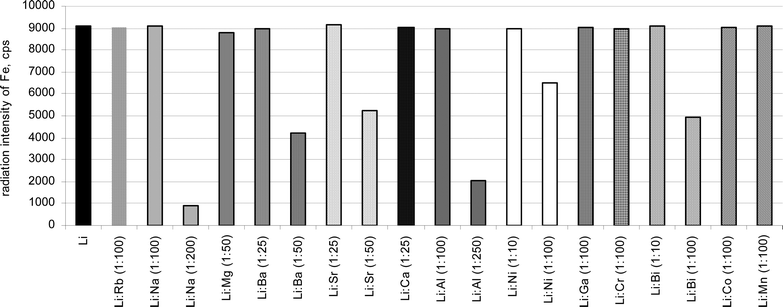 | ||
| Fig. 2 Effect of sodium, rubidium, aluminium, alkaline-earth metals and selected trivalent 3d transition-metal ions on the precipitation of potassium lithium periodatoferrate(III). | ||
The evaluation of the influence of trivalent 3d transition-metal ions on the precipitation of the LiKFe(IO6) complex were also investigated. Attempts were made to incorporate Ni, Ga, Cr, Bi, Co and Mn. The obtained results indicate that gallium and chromium do not precipitate in the applied conditions. Thus, these elements cannot affect Fe Kα and the determination of lithium. A trace amount of bismuth and manganese was found in analyzed precipitates. Carried out experiments indicate also that some amounts of nickel and cobalt are present in the precipitates. The radiation intensities of Ni Kα and Bi Kα are the same both in the synthetic samples (with lithium) and blank samples and these values increase along with increasing added amounts of the elements. Thus, the net counts per second for these elements are close to zero. In the case of the remaining elements, the emitted radiation is more intensive in the synthetic samples with lithium than in the blank samples. The potassium lithium periodatoferrate(III) complex can be the carrier for the above mentioned elements, thus, the radiation of Co Kα and Mn Kα is more intensive in the samples with lithium than in the samples without lithium. Nevertheless, the amount of cobalt and manganese coprecipitated with the potassium lithium periodatoferrate(III) complex do not affect the radiation intensity of iron, even if the ratio of the added elements to lithium is 100:1 (5 mg Co or Mn). In the case of nickel and bismuth, their influence on iron appears when the ratio of the added elements to lithium is 100:1 (5 mg Ni or Bi). The lower concentration of nickel and bismuth does not interfere with the indirect determination of lithium. The radiation intensity of iron, independently of the presence of the mentioned elements, is not changed. The attempts to form analogues of LiKFeIO6 with Cr3+, Ni3+ or Mn3+ instead of Fe3+ have failed. The Co3+ can form a similar complex, but in this case different, more restrictive conditions of precipitation are required.18
The self-absorption effect was evaluated with the formula of the absorption correction factor calculated using the emission–transmission (E-T) method. This method involves measuring of the Fe radiation in the precipitate collected on the Millipore filters without a target (pellet of Fe2O3), with a target located at a position adjacent to the back of the filters and the target covered the filter without precipitate. The measured radiation intensity of iron in potassium lithium periodatoferrate(III) of various masses was corrected using the absorption correction factor calculated from equations: Hi = exp[-χi(λ,λi)m] = (IT + S,i – IS,i)/IT,i and Ai = (1- exp[-χi(λ,λi)m])/[-χi(λ,λi)m; where: IT + S,i, IS,i, IT,i are the net intensity from the sample plus target, from the sample alone and from the target alone (with Millipore filter), respectively; Ai is the absorption factor, χ(λ,λi) is the total mass-attenuation coefficient for incident λ and fluorescent radiation λi in cm2 g−1, m is the mass per unit area of the sample in g cm−2. The change of obtained absorption factors for samples with different amounts of lithium collected onto the Millipore filters are presented in Table 1. The values of the absorption factors decrease along with the increase of the mass per unit area of the samples. Thus, the investigation shows that the obtained results have to be corrected by the absorption factors. Fig. 3 shows the radiation intensity of the iron in loaded potassium lithium periodatoferrate(III) before and after absorption correction. Table 2 presents the calibration parameters (least-squares method), the correlation coefficients, r, and residual errors, RMS (root of the mean square of the sum of the differences between the measured values and the calculated values), before and after absorption correction. The values of r and RMS indicate the considerable improvement in the results after the correction of matrix effects (self-absorption effects and sharply increasing effects with the mass per unit area of collected material) using absorption factors.
| Li in LiKFeIO6 complex, (µg) | Mass per unit area, (µg cm−2) | Absorption factor |
|---|---|---|
| 0 | 0 | 0.999 |
| 20 | 189 | 0.950 |
| 30 | 284 | 0.931 |
| 40 | 379 | 0.910 |
| 50 | 473 | 0.881 |
| 60 | 568 | 0.866 |
| 70 | 662 | 0.840 |
| Mass range of Li, (µg) | Before matrix correction | After matrix correction | ||
|---|---|---|---|---|
| R | RMS | R | RMS | |
| 20–70 | 0.9917 | 3.399 | 0.9994 | 0.926 |
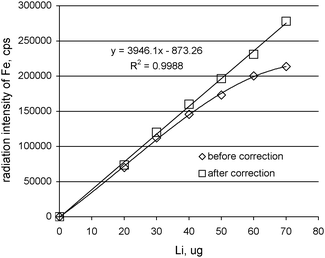 | ||
| Fig. 3 The results for lithium standards before and after absorption correction. | ||
The precipitation yield of lithium in both synthetic and Standard Reference Materials of Lithium Ores was evaluated by using the following formula: R = ((madded – msolution)/madded)·100%; where: madded is the mass of lithium (in µg) introduced into the solution before the precipitation of potassium lithium periodatoferrate(III) complex and msolution is the mass of lithium (in µg) remaining in the solution after the precipitation of the mentioned complex. The detailed results are given in Table 3. The precipitation yield of lithium equals ca. 98.5% independently of the kind of the sample i.e. synthetic standards and SRM ores. Not only good recovery but also repeatability of the analysis results were achieved for homogeneously loaded potassium lithium periodatoferrate(III) complex over the whole area of the membrane filter. The repeatability of sample preparation has been investigated on the basis of seven samples of standards and seven samples of SRM lithium ores, which were specially prepared for this purpose. The results are presented in Table 4. The obtained relative standard deviations of ca. 3% for both synthetic standard and SRM 182 standard can be recognized as satisfactory.
| Sample | Added, (µg) | Determined in filtrate, (µg) | Recovery, (%) |
|---|---|---|---|
| Synthetic Standard | 30 | 0.390 | 98.7 |
| SRM 182 | 30 | 0.450 | 98.5 |
| SRM 181 | 45 | 0.675 | 98.5 |
| Sample, ca. 30 µg Li | Standard deviation, (µg) | Relative standard deviation, (%) |
|---|---|---|
| Synthetic Standard | 0.930 | 3.13 |
| SRM 182 | 0.862 | 2.88 |
The detection limit is dependent on the amount of lithium in the potassium lithium periodatoferrate(III) complex that can be precipitated. The proposed method allows the minimum precipitation of 20 µg. But XRF measurements and the use of the very sensitive Fe Kα line allow the determination of lower amounts of lithium. When the detection limit is calculated from DL = (3/k)(B/t)1/2, where k is the sensitivity in counts s−1 µg−1, B is the background count rate in counts s−1 and t is the counting time, the obtained DLs with a counting time of 20 s can be equal to 14 ng of lithium (t = 20 s) or even 6 ng if the time of measurement equals 100 s.
Table 5 presents the accuracy of the XRF analyses based on the measurement of reference materials. The standard reference materials of lithium ores no. 181 and 182 with certified lithium concentrations equal to 6.39% and 4.34%, respectively, were tested. The obtained concentrations from the XRF analysis were 6.52% and 4.24% for these materials. Thus, the relative errors of the mentioned analysis were 2.03% and 2.25%, respectively. The agreement between XRF analysis and certified values is satisfactory and indicates the usefulness of the presented method for the determination of lithium in lithium ores.
| Sample | WDXRF analysis, µg | Certified value, µg | Relative error, % |
|---|---|---|---|
| SRM 181 | 45.44 ± 0.269 | 44.53 | 2.04 |
| SRM 182 | 29.56 ± 0.172 | 30.24 | 2.25 |
Conclusions
An indirect method for the determination of minor amounts of lithium by XRF spectrometry using stoichiometric potassium lithium periodatoferrate(III) complex was developed. In the proposed method, lithium is determined via XRF measurement of iron in the above mentioned complex with a good ratio of lithium to iron (1 to 8). The main advantage of the proposed method is that it makes it possible to determine a very light element, undetectable by commercially available XRF spectrometers due to the extremely low fluorescent yield and long-wavelength characteristic radiation. The selected elements do not directly influence the precipitation of the potassium lithium periodatoferrate(III) complex and the determination of lithium by XRF measurement of iron. The samples were prepared with RSD ≈ 3% for small amounts of lithium (30 µg). The developed method is not only selective and precise but also accurate (relative error ca. 2%). The method can be applied inter alia to the determination of the trace amounts of lithium in the lithium ores. Further work is in progress and it will deal with the extension of the method and the determination of lithium in various materials.Acknowledgements
The author acknowledges Maria Czaja and Tomasz Krzykawski from the Department of Earth Sciences, Silesian University in Sosnowiec, for their kind collaboration.Notes and references
- D. H. Loring, Mar. Chem., 1990, 29, 155–168 CrossRef CAS.
- H. Okamoto, Y. Okamoto, T. Hirokawa and A. R. Timerbaev, Analyst, 2003, 128, 1439–1442 RSC.
- N. Jabeen, E. Rehman and S. Ahmed, J. Radioanal. Nucl. Chem., 2003, 258, 427–430 CrossRef CAS.
- U. Nickus and H. Thies, J. Chromatogr., A, 2001, 920, 201–204 CrossRef CAS.
- R. P. Singh and N. M. Abbas, J. Chromatogr., A, 1996, 733, 93–99 CrossRef CAS.
- H. Hamzaoui, A. M'nif and R. Rokbani, Talanta, 2006, 70, 847–851 CrossRef CAS.
- R. O. Bach, Lithium Current Application in Science, Medicine and Technology, Wiley, New York, 1985 Search PubMed.
- W. Mertz, 1986, In: ed. W. Mertz, Trace Elements in Human and Animal Nutrition, Vol. 2. Academic Press, Orlando, FL, pp. 391–397 Search PubMed.
- B. Baraj, L. F. H. Niencheski, R. D. Trapaga, R. G. Franca, V. Cocoli and D. Robinson, Fresenius J. Anal. Chem., 1999, 364, 678–681 CrossRef CAS.
- P. Lefton, R. Plaquet, F. Rose, G. Hennon and N. Ledeme, Anal. Chim. Acta, 1996, 327, 301–306 CrossRef CAS.
- M. Taddia, P. Modesti and A. Albertazzi, J. Nucl. Mater., 2005, 336, 173–176 CrossRef CAS.
- M. F. S. Teixeira, M. F. Bergamini and N. Bocchio, Talanta, 2004, 62, 603–609 CrossRef CAS.
- M. Cretin, L. Alerm, J. Bartroli and P. Fabry, Anal. Chim. Acta, 1997, 350, 7–14 CrossRef CAS.
- M. Tabata, J. Nishimoto and T. Kusano, Talanta, 1998, 46, 703–709 CrossRef CAS.
- C. L. Luke, Anal. Chim. Acta, 1969, 45, 365–376 CrossRef CAS.
- O. Procke and A. Slouf, Coll. Czech. Chem. Commun., 1939, 11, 273 CAS.
- W. A. Nazarenko and W. Filatowa, J. Anal. Chim., 1950, 5, 234–237 Search PubMed.
- D. Currie, A. Hector, W. Levason and M. Thomas, J. Mater. Chem., 1997, 7, 1871–1875 RSC.
| This journal is © The Royal Society of Chemistry 2010 |