Templated nanoscale porous carbons
Yongde
Xia
,
Zhuxian
Yang
and
Robert
Mokaya
*
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, United Kingdom. E-mail: r.mokaya@nottingham.ac.uk
First published on 26th February 2010
Abstract
This manuscript reviews key developments in the important and rapidly expanding area of templated porous carbons. The porosity covered ranges from microporous to mesoporous and macroporous carbons. Two modes of templating, using so-called hard and soft templates, are covered. In particular, for hard templating, zeolite templating generates microporous carbons, mesoporous silicates yield mesoporous carbons, while colloidal particles are replicated to large mesoporous and macroporous carbons. Soft-templating, a more recent phenomenon, mainly generates mesoporous carbons. The full range of pore sizes can therefore now be accessed using hard and soft templates to generate highly ordered nanoscale carbons with well-defined and optimised textural properties. The research area has seen rapid and important developments over the last few years, and this review aims to present the more significant advances.
 Yongde Xia | Yongde Xia obtained his PhD in 1999 from Fudan University in China, followed by postdoctoral research at Korea Advanced Institute of Science and Technology and the University of Paris-Sud. He moved to the United Kingdom in 2001 and worked as a research fellow with Prof. Robert Mokaya in the School of Chemistry at the University of Nottingham. He is now a research fellow in the School of Mechanical, Materials and Manufacturing Engineering at the University of Nottingham, working on the applications of porous materials in catalysis and energy-related gas storage. |
 Zhuxian Yang | Zhuxian Yang received her BSc from East China Normal University in 1995 and her MSc from Fudan Univeristy, China in 1998. She earned her PhD in 2007 from the School of Chemistry at the University of Nottingham in the United Kingdom, under the supervision of Prof. Robert Mokaya, working on the synthesis, characterisation and application of micro/mesoporous carbon materials. She is now a research fellow studying the hydrogen storage of multi-component metal hydrides and confined hydrides in the School of Mechanical, Materials and Manufacturing Engineering at the University of Nottingham. |
 Robert Mokaya | Robert Mokaya received his BSc degree in chemistry from the University of Nairobi, Kenya in 1988. He obtained a PhD from the Department of Chemistry, the University of Cambridge (with Professor William Jones) in 1992. He was elected a Research Fellow at Trinity College, Cambridge (1992–1996), and awarded an EPSRC Advanced Fellowship (1996–2001). Currently, he is Professor of Materials Chemistry in the School of Chemistry, the University of Nottingham. His research interests include the design, synthesis and characterisation of novel porous inorganic and carbon materials and the study of their structure–property relations. |
1. Introduction
Porous materials contain voids as the majority phase, either with random character (disordered pore systems) or with high regularity (ordered pore systems).1 Ordered porous materials are of great scientific and technological interest due to their ability to interact with atoms, ions and molecules not only at their surfaces, but also throughout their bulk.2 The pore system of any porous material may be classified, according to IUPAC (International Union of Pure and Applied Chemists), based on the pore diameter. Pores below 2 nm are classified as micropores and the typical representative materials are microporous zeolites. Pores that fall in the size range between 2 and 50 nm are called mesopores and, the mesoporous M41S family and SBA series of materials are prominent examples of this class. Pores above 50 nm are called macropores, and amorphous aluminosilicates and porous glass are typical representatives of macroporous materials.Porous carbons, a class of non-oxide porous materials, are of great importance due to their applications in water and air purification, as gas hosts, templates,3,4 or components of electrodes.5 The widespread use of porous carbons results from their remarkable properties, such as hydrophobicity of their surfaces, high surface area, large pore volume, chemical inertness, good thermal stability, good mechanical stability, easy handling and low cost of manufacture.6 Most porous carbons are primarily microporous and the microporous nature is well-suited to many applications involving small molecules, such as molecular sieving, adsorption and catalysis.7,8 However, there are a number of other potential uses in which the presence of mesopores or even macropores would be preferable, for instance, adsorption of large hydrophobic molecules such as vitamins, dyes and polymers, chromatographic separations or in electrochemical double-layer capacitor applications.7 Therefore, the development of mesoporous and macroporous as well as microporous carbon materials is of great importance not only from a fundamental research point of view, but also from the practical application point of view.
Porous carbons are usually obtained via carbonisation of precursors of natural or synthetic origin, followed by activation.9 Various synthesis strategies have been explored to prepare porous carbons with controlled pore structures both at the micropore and mesopore level.7 At the micropore level, molecular sieving carbons that possess uniform micropores may be prepared via carbonisation of appropriate carbon precursors. For example, Miura et al. illustrated the potential of targeted synthesis of molecular sieving carbons with a pore size of ca. 0.35 nm by changing the carbonisation temperature and the mixing ratio of coal, pitch, phenol and formaldehyde precursors.10 Microporosity could also be controlled by carbonising ion-exchange resin at 500–900 °C, whereby spherical polystyrene based resins with a sulfonic acid group were ion-exchanged with several kinds of cations including H+, K+, Na+, Zn2+, Cu2+, Fe2+, Ni2+ and Fe3+.11 The molecular sieving carbons prepared from the resins with di- or trivalent cations had pore size varying between 0.38 and 0.45 nm depending on the type of cation.11 Recently, microporous carbons with pore size distribution in the range of 0.77–0.91 nm were obtained via carbonisation and activation of waste-derived biomass.12 The biomass-derived microporous carbons exhibit high surface area of up to 3100 m2 g−1, large pore volume of up to 1.68 cm3 g−1 and considerable gas uptake capability. In the mesopore regime, catalytic activation,13,14 and carbonisation of polymer blends,15 organic gels,16 and colloidal imprinting,17–19 have been proposed for the preparation of mesoporous carbons. In general, mesoporous carbon materials made from catalytic activation and carbonisation usually display disordered structures with wide pore size distribution.13–16 This is because the pores in such disordered porous carbons are generated by etching processes that are difficult to control or by escaping gases during carbonisation of carbon precursors.
To improve the level of structural ordering, a template can be used to guide the formation of pores during the carbonisation process. Template carbonisation routes allow the preparation of carbon materials with controlled architecture and relatively narrow pore size distribution.20 Therefore, template carbonisation has attracted much attention for the preparation of ordered porous carbons. Generally, two types of templates, classified as soft template or hard template, are used as molds to form porous materials. The hard template carbonisation approach, as shown in Fig. 1,21 usually involves the following steps: (a) the preparation of a porous template with controlled porosity; (b) the introduction of a suitable carbon precursor into the template pores either by wet impregnation, chemical vapour deposition or a combination of both methods; (c) the polymerisation and carbonisation of the carbon precursor to generate an organic–inorganic composite; and (d) the removal of the inorganic template to yield a porous carbon. Following this template procedure, the carbon formed in the pores of the host template turns into a continuous carbon framework while the space once occupied by the host template is transferred into the pores of the resulting carbon material. Hard templates therefore offer rigid nanocast molds as true templates to replicate other materials. The hard templates must be thermally stable, chemically inert to carbon precursors and be amenable to removal to generate a pure carbon framework. Ordered inorganic porous solids are the mostly common used hard templates. It is worthwhile pointing out that the pore size of the resulting porous carbon materials is not always exactly the size of the pore walls of host templates since there is some shrinkage during carbonisation, which is usually carried out at elevated temperatures.
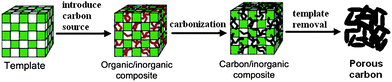 | ||
| Fig. 1 Schematic illustration of the hard template method for the preparation of porous carbon materials from porous inorganic templates. Reproduced with permission from ref. 21. | ||
On the other hand, the soft template method involves cooperative assembly between the structure-directing agents (usually surfactants or copolymers) and organic precursor species in solution. Therefore the carbon structures obtained via soft templates are more flexible and their formation is dependent on temperature, type of solvent and ionic strength. However, there are currently only limited examples of the successful fabrication of porous carbon via soft templating methods, which were reviewed recently by Zhao and colleagues.22 Such templating of porous carbon can be considered as being endotemplating, whereby the templating species are added to the synthesis mixture and are occluded in the growing carbon framework and generate a pore system in the carbon after their removal.22–24 This contrasts with exotemplating where the templates are materials with structural pores in which the carbon framework is created. The exotemplate therefore acts as a scaffold for the formation of a carbon framework and on removal generates a porous carbon.23,24
Historically, the hard template carbonisation method was first reported by Knox and co-workers in 1986,25 who demonstrated the synthesis of graphitic porous carbons for liquid chromatography separation by impregnation of spherical porous silica gel particles with phenolic resin and subsequent carbonisation and removal of silica. Since this first report, the method has been extensively employed to prepare ordered porous carbons.20,26–28 A variety of inorganic templates including zeolites,28–31 mesoporous silicas,5,20,26,32–37 colloid crystals,38,39 poly(styrene)40 and aluminium oxide membranes have been explored to synthesise microporous, mesoporous and macroporous carbon materials. Various carbon precursors including sucrose,26 furfuryl alcohol,28,29,41–43 acrylonitrile,28,42 propylene,29,30 pyrene,42 vinyl acetate,42 and acetonitrile33,44–46 have been used to prepare porous carbons. Indeed, the past decade has witnessed rapid advances in the use of template carbonisation to produce ordered porous carbon materials, ranging from microporous to mesoporous and macroporous carbons. The template carbonisation method has thus been regarded as one of the most effective approaches to prepare porous carbon materials with desirable physical and chemical properties. It has therefore opened new opportunities in making novel porous materials for a wide range of applications.
Several reviews covering the synthesis, properties and applications of porous carbons, especially mesoporous carbon materials can be found in the literature.47–55 In this review, we summarise the early and recent developments in the synthesis and characterisation of templated porous carbon materials. Particular attention is paid to the synthesis of structurally well ordered porous carbon materials with narrow pore size distribution via both hard template and soft template methods. We especially emphasise those so-called breakthroughs in the preparation of porous carbon materials. This review is divided into three sections according to the pore size of carbon materials: we first consider the synthesis of microporous carbon materials using zeolites and clays as hard templates, then summarise the preparation of mesoporous carbon materials via both hard template and self-assembly soft template methods, followed by presentation of the preparation of macroporous carbon materials from crystal colloid templates, and finally we offer some concluding remarks on the development of nanoscale porous carbon materials.
2. Microporous carbon materials
2.1. Zeolites as hard templates
Zeolites are highly crystalline aluminosilicate materials that possess uniform sub-nanometre sized pores. The pore channel apertures vary between 0.3 and 1.5 nm,56 depending on the type of zeolite and its preparation history, e.g. calcination, leaching or various chemical treatments. Zeolites have been widely used in ion exchange, separation, catalysis, chemical sensing and host–guest chemistry, particularly as shape-selective catalysts owing to their uniform molecular-sized pores.57 Since the wall thickness of zeolites is less than 1 nm, the periodic pore structure and well-defined internal nanospaces of zeolites provide excellent opportunities to control the nanostructure and morphology of microporous carbon materials at the nanometre level. It is reasonable to expect that if such a nanospace in a zeolite is filled with carbon, the generated porous carbon should reflect the porosity of the original zeolite template. Furthermore, the porous carbons replicated from zeolites should possess a narrow pore size distribution due to the uniformity of the zeolite pore wall thickness. Narrow micropore size distribution is an obvious advantage of zeolite-templated carbons over conventional activated carbons which usually possess wide pore size distributions in both the micropore and mesopore size range. Zeolites have therefore been widely used as inorganic templates for the synthesis of microporous carbons with uniform pore size.28–30,42,43,58–62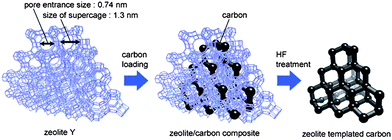 | ||
| Fig. 2 Scheme for the synthesis of microporous carbons templated from zeolite Y. Reproduced with permission from ref. 63. | ||
The studies mentioned above, on zeolite-templated carbonisation, exclusively resulted in disordered microporous carbons with a significant proportion of mesopores.28 This means that zeolite-type structural ordering was not faithfully transferred to the carbons.28,64 This could be due to low carbon precursor loading and/or spatial restrictions (within the zeolite template pores) during the precursor polymerisation58 and limited subsequent carbon growth.28 A consequence of such incomplete carbon infiltration and partial filling of the zeolite pores is that the internal porosity of the resulting carbons is not a true replica of zeolite-like ordering even though the morphology of the carbons is similar to that of the zeolite template. The generation of mesopores in such disordered zeolite templated carbons results from the partial collapse of the carbon framework after the removal of the zeolite framework. The collapse may be explained by considering that the thin walls of the generated carbon, which are derived from the partially filled pores of the zeolite template, do not exhibit sufficiently high mechanical strength to survive removal of the zeolite framework.
2.1.2.1. Zeolite Y as a hard template. In order to improve the structural ordering of zeolite templated carbons, Kyotani and co-workers systematically investigated the synthesis of microporous carbons using zeolite Y as a hard template. A two-step method was used to prepare ordered microporous carbon with high surface area, which retained the structural regularity of zeolite Y, by filling as much carbon precursor as possible into the zeolite pores so as to prevent any subsequent partial collapse of the resulting carbon framework.31 In the two-step method, additional incorporation of carbon was achieved by a chemical vapour deposition (CVD) process using propylene gas as the carbon source after an initial carbonisation step involving the heat treatment of a zeolite/furfuryl alcohol composite at 700 °C. The obtained carbon after the removal of the zeolite template exhibited an ordered zeolite replica structure with long-range ordering, confirmed by a (111) reflection (similar to that of zeolite Y) at 2θ = 6.26° in the XRD pattern, as shown in Fig. 3. This was the first report on the successful (according to XRD patterns) replication of zeolite-type ordering from a zeolite template to microporous carbon. However it is worth noting that although ordered microporous carbon materials with a negative replica structure of zeolite Y were obtained by this two-step method, there was still an amorphous (002) peak at 2θ = 23° in the XRD pattern, which indicated the partial collapse of the carbon framework and/or the presence of graphitic/turbostratic domains deposited outside the zeolite pore system.
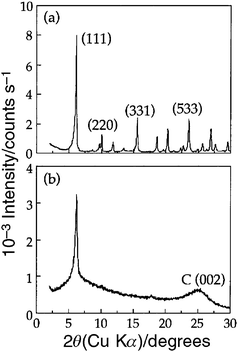 | ||
| Fig. 3 XRD patterns of (a) zeolite Y and (b) zeolite Y templated carbon.31 | ||
Kyotani and co-workers improved the synthesis of ordered microporous carbon by heat treatment of the carbon/zeolite composite obtained by the two-step method at 900 °C.65 The carbon seemed to be better carbonised and had a more rigid and stable structure with enhanced replication of zeolite-like ordering, and exhibited a high surface area of up to 3600 m2 g−1 and pore volume of 1.5 cm3 g−1. Fig. 4 shows a high-resolution transmission electron microscopy (HRTEM) image of the ordered microporous carbon obtained from zeolite Y.65 Kyotani and co-workers extended the two-step replication process to other zeolite templates including zeolite beta, ZSM-5, mordenite and zeolite L, and obtained a variety of ordered microporous carbon arrays.29 The optimum conditions used to obtain carbons with the highest long-range ordering varied depending on the zeolite template used. The order of level of zeolite-like structural regularity in the templated carbons was zeolite beta ≫ zeolite L > mordenite > ZSM-5. The trends in ordering of the carbons suggested that in order to obtain microporous carbon with high structural regularity, the zeolite template should have a larger pore channel (>0.6–0.7 nm), and at the same time, the channel system should be three-dimensionally interconnected.29 Kyotani and co-workers also prepared microporous carbons with zeolite-type structural ordering and BET surface area exceeding 4000 m2 g−1via CVD procedures that excluded an initial liquid impregnation step.66 It was claimed that low-temperature CVD followed by high-temperature thermal treatment is essential to produce ordered porous carbon materials.66 Nitrogen-containing zeolite Y-templated microporous carbon with good structural ordering has also been prepared by combining liquid impregnation of furfuryl alcohol and CVD of acetonitrile.46
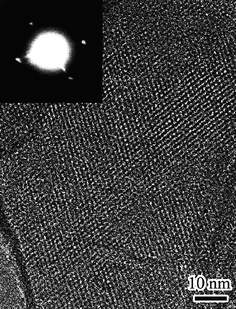 | ||
| Fig. 4 HRTEM image and corresponding electron diffraction pattern (inset) of zeolite Y templated carbon. Reproduced with permission from ref. 65. | ||
Several other groups have presented interesting results on the preparation of ordered zeolite templated carbons. Garsuch and Klepel reported on the preparation of microporous carbons that preserved the structural regularity of the zeolite Y templates and found that the quality of the carbons strongly depended on heat treatment of the zeolite/carbon composite prior to etching.61 Furthermore, the particle size of a zeolite template also affected the structural regularity of the resulting carbon materials.62 This may be understood by considering that particle size affects the diffusion of carbon precursor into the zeolite pores: small particles allow more extensive infiltration of the zeolite template pores and therefore favour the formation of ordered microporous replica carbons.62 Barata-Rodrigues and co-workers found that the use of zeolite templates imparted controllable features to the replica carbon, which could not be achieved by activated carbon preparation methods.60 Interestingly, additional porosity could be introduced to the replica carbons by controlled carbonisation and demineralisation or by incomplete filling of the template's pore network.60 Microporous carbon with a crystallographically amorphous carbon core and graphitic carbon shell was prepared by Zhao and co-workers using proton exchanged zeolite Y as the template, and it was observed that a Pt catalyst supported on the replica carbon had a higher specific activity for room-temperature methanol oxidation than a commercial catalyst consisting of Pt deposited on Vulcan XC-72 carbon black.67 Béguin and co-workers reported the synthesis of zeolite Y templated carbon doped with nitrogen by a two-step nanocasting process using acrylonitrile and propylene as precursors.68 The carbon inherited the ordered structure of the zeolite Y template and had a narrow pore-size distribution within the micropore range, and a large number of heteroatoms including both oxygen and nitrogen. The templated carbon material was found to display a large gravimetric capacitance of 340 F g−1 in aqueous media because of the combined electrochemical activity of the heteroatoms and the accessible porosity. This N-doped carbon replica could operate at 1.2 V in aqueous medium with good cycling ability (i.e., beyond 10000 cycles), and is extremely promising for use in high-energy-density supercapacitors.
2.1.2.2. Other zeolites as hard templates. Using a two-step method, Gaslain and co-workers have reported the preparation of a porous carbon replica with a well resolved X-ray diffraction pattern using the wider pore channel zeolite EMC-2 as a template.69 The improved structural regularity of the replicated carbons was demonstrated by three well resolved XRD peaks corresponding to (100), (002) and (101) reflections of the EMC-2 zeolite, as shown in Fig. 5. The resulting carbon materials display high surface area and pore volume without any significant contribution from mesoporosity, possibly due to enhanced carbon precursor infiltration resulting from the presence of a straight pore channel system in the zeolite EMC-2 template. Other zeolites that have been used as templates include zeolite LTA (from which a nanostructured porous carbon was obtained using CVD of methanol as the carbon source),70 zeolite MCM-22 (with sucrose as the carbon precursor)71 and natural zeolites (with sucrose as the carbon precursor).72
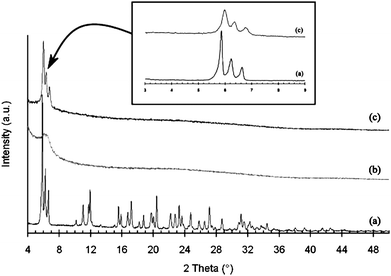 | ||
| Fig. 5 XRD patterns of: (a) zeolite EMC-2, (b) a carbon replica prepared by furfuryl alcohol infiltration and propylene CVD and (c) a carbon replica prepared by furfuryl alcohol infiltration, propylene CVD and heat treatment at 900 °C under argon.69 | ||
Mokaya's group has also extensively explored the use of zeolites as templates to synthesise microporous carbon materials.45,73–76 Using zeolite beta or silicalite-I as templates and acetonitrile as the carbon source (via CVD), hollow shells of porous nitrogen-doped carbon materials with high surface area can be produced.45 The carbon materials generally retain the particle morphology of the zeolite templates. However, when CVD is performed at temperatures ≥ 900 °C, hollow carbon shells that are hexagonal, cubic, or rectangular in shape are obtained as the predominant particle morphology, as indicated in Fig. 6. Carbon materials prepared below 950 °C with zeolite beta as a template have a high surface area of up to 2270 m2 g−1 and contain significant amounts of non-graphitic (i.e., amorphous) carbon that exhibits structural pore channel regularity replicated from the zeolite. It is thus possible by choice of zeolite template and CVD conditions to nanocast nanoporous carbon materials that exhibit a hollow-core particle morphology, high surface area and zeolite-type pore channel ordering or hollow shells with significant levels of graphitisation.45
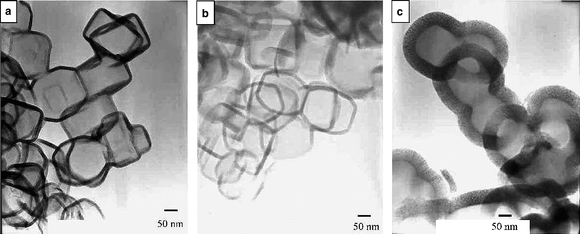 | ||
| Fig. 6 TEM images of carbon materials prepared via CVD using silicalite-I as the template at various CVD temperatures: (a) 900 °C, (b) 950 °C and (c) 1000 °C.45 | ||
Porous carbon materials may also be prepared using zeolite 13X as a template and acetonitrile or ethylene as the carbon source via CVD at 550–1000 °C.73 Carbons obtained from acetonitrile have a high surface area of up to 1920 m2 g−1, a high pore volume of up to 1.4 cm3 g−1, and exhibit some structural ordering replicated from the zeolite template. When ethylene is used as a carbon precursor, high surface area (1300 m2 g−1) carbons are only obtained at a lower CVD temperature of 550–750 °C. The ethylene-derived carbons retain some zeolite-type pore channel ordering but also exhibit significant levels of graphitisation even at low CVD temperatures. Such zeolite-templated carbons have a hydrogen uptake of up to 4.5 wt% at −196 °C and 20 bar, with uptake strongly dependent on surface area.73 Evaluation of the performance of the templated carbons in electrochemical capacitors with tetraethylammonium tetrafluoroborate (NEt4BF4) salt solution in acetonitrile showed a high gravimetric capacitance of up to 146 F g−1 and a short time constant of up to 3 s, indicating a fast charge/discharge capability. Specific capacitance at a constant pore size was found to linearly depend on the surface area. The electrochemical data show that carbons produced by zeolite templating are promising for use in high-energy, high-power electrochemical capacitors employing organic electrolytes.77
Several studies have shown a link between the type of zeolite template and the level of zeolite-like ordering achieved in the replica carbon. The use of large pore zeolites with a two- or three-dimensional non-cubic pore system (such as zeolite EMC-2) was suggested as being beneficial for nanocasting highly ordered zeolite templated carbons. However, Mokaya and co-workers have nevertheless demonstrated the synthesis of structurally well ordered zeolite-like carbons using zeolite beta.74 Furthermore, the zeolite-like carbons were obtained via simple CVD rather than a combination of liquid impregnation and CVD.74 The zeolite-like carbons were prepared via CVD at 800 or 850 °C using acetonitrile as the carbon precursor, and exhibited XRD patterns with at least two well-resolved diffraction peaks and TEM images that revealed well-ordered micropore channels, as shown in Fig. 7. The carbons possess a surface area of up to 3200 m2 g−1 and pore volume of up to 2.41 cm3 g−1. A significant proportion (up to 80%) of the porosity in the carbons is from micropores, and the porosity is dominated by pores of size 0.6–0.8 nm, which show an enhanced hydrogen storage capacity of up to 6.9 wt%.74 Moreover, isosteric heat of adsorption of up to 8.2 kJ mol−1 for the zeolite-like carbons indicated a strong interaction between adsorbed hydrogen and the carbon surface. Pacula and Mokaya subsequently used as-synthesised zeolite beta as template for the preparation of well ordered zeolite-like carbon materials that also exhibit a high surface area (up to 2500 m2 g−1) and pore volume (up to 1.56 cm3 g−1), along with high hydrogen uptake capacity.75 The use of as-synthesised rather than calcined zeolite beta significantly improved the carbon yield and reduced the number of steps in the preparation of the templated carbons. More recently, well ordered and high-surface-area microporous carbons that are N-doped or N-free were fabricated using zeolite EMC-2, 13X or Y as templates. The carbons were used to experimentally demonstrate that N-doping can remarkably influence the hydrogen uptake capacity of high-surface-area microporous carbons.76
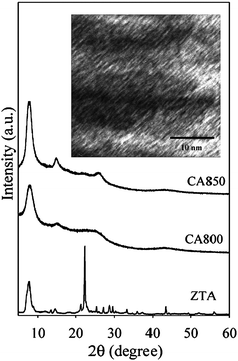 | ||
| Fig. 7 Powder XRD patterns of zeolite beta template (ZTA) and corresponding carbon materials (CA800 and CA850) prepared at CVD temperatures of 800 and 850 °C respectively. The inset shows a representative TEM image of sample CA850.74 | ||
2.2. Clays as hard templates
Clays have also been explored as templates for the synthesis of porous carbon materials. Carbonisation of organic polymers in the interlayer region of a layered clay can produce thin graphite films. A typical non-graphitisable carbon precursor like poly(furfuryl alcohol) can be readily graphitised by using layered clays as hard templates. This is somewhat beyond the bounds of the conventional common knowledge of carbon science, where it is considered that the final structure of a carbon material strongly depends on the nature of the original precursor rather than the conditions of the carbonisation process.Kyotani and co-workers have prepared carbon materials using montmorillonite clay as template and poly(acrylonitrile), poly(furfuryl alcohol) and poly(vinyl acetate) as carbon precursor.78–80 They found that the carbon precursors were easily graphitised to generate carbon as two-dimensional film-like graphite, consisting of highly oriented layer planes which were stacked thin and wide.79,80 Porous carbons may also be prepared from poly(furfuryl alcohol) using various layered clays, including taeniolite and saponite, as templates.81 Such carbons exhibit a film-like, highly stacked structure of (002) planes and are highly graphitised. Bandosz et al. synthesised carbon materials via carbonisation of furfuryl alcohol within a sodium form of Wyoming smectite clay intercalated with hydroxyaluminium cations, and concluded that the inorganic matrix acts as a limiting pore size former.82 Bandosz and co-workers also obtained carbons by carbonisation of poly(furfuryl alcohol) within smectite and taeniolite matrices, and found that the resulting carbon materials presented sieving effects for molecular sizes between 3.6 and 6 Å.83 The lithium form of taeniolite, intercalated with hydroxyaluminium and hydroxyaluminium-zirconium cations also served as template to fabricate carbon materials.59 The work of Bandosz and co-workers demonstrated that the structural properties of the carbon materials are also affected by the water content of the template inorganic matrix. Using a Na-taeniolite clay film as hard template and poly(acrylonitrile) as the carbon precursor, a flexible graphite film with crystallised and oriented graphite characteristics can be prepared.84 Commercial cationic bentonite clay was used as an inorganic template with furfuryl alcohol together with an additional treatment of propylene vapour deposition, as a carbon source to generate rather poorly ordered carbon with a relatively low surface area of 446 m2 g−1.60 The carbon produced with additional propylene showed more densely packed crystals than the carbon produced from furfuryl alcohol impregnation only, which could be an indication of a stronger carbon network left after removal of the template. Mg–Al layered double hydroxides intercalated with 1,5-naphthalene disulfonate dianions have also been carbonised to produce carbon materials that exhibit heterogeneous micropore structures with pore diameters no larger than the interlayer space of the clay.85
Pillared clays (i.e., layered silicates whose sheets have been permanently propped open by thermally stable molecular props), were used as templates to generate porous carbons with pore diameters ranging from 0.8 to 2.2 nm.86 An approach to pyrolyse aromatic hydrocarbons such as pyrene within a pillared clay was also reported, in which the pillared clay serves two functions.87 It acts as the inorganic template around which the carbon can be formed, and it also functions as an acid catalyst to promote condensation of the aromatics in a manner similar to the Scholl reaction. The resulting carbon materials have pore sizes from 1.5 to 5 nm.87
Recently, Mokaya's group has reported that porous carbon materials can be prepared using Mg–Al layered double hydroxides as templates via CVD using acetonitrile as the carbon precursor.88 Depending on the CVD temperature, the carbons exhibit microporosity and/or mesoporosity and significant levels of graphitisation. The layer structural ordering of the layered double hydroxides is somewhat retained in the layered double hydroxide/carbon composite obtained after CVD, but is lost after removal of the template to generate carbon. The morphology of the layered double hydroxide template is retained in the carbon materials, which in general consist of an assembly of small flaky particles.
2.3. Other microporous materials as hard templates
Apart from zeolites and clays, other materials, such as metal–organic frameworks (MOFs), have also been explored as template to produce porous carbons. Recently, Liu and co-workers have synthesised porous carbon by heating the carbon precursor furfuryl alcohol within the pores of MOF-5 followed by thermal treatment for carbonisation and removal of the MOF template. The MOF-templated carbon exhibits a high surface area of up to 2872 m2 g−1 and a high pore volume of 2 cm3 g−1, but possesses both micropores and mesopores. The presence of mesopores may be due to the formation of voids between particles or incomplete infiltration of carbon precursor into the MOF pore channels. The MOF-templated carbon was found to exhibit good hydrogen uptake as well as excellent electrochemical properties as an electrode material for electrochemical double-layered capacitors.892.4 Overview and perspective
The past decade has seen significant advances in the synthesis of ordered microporous carbons. The use of zeolites as hard templates has in particular provided porous carbon materials with interesting textural properties. The emergence of the so-called zeolite-templated carbons (ZTCs) or zeolite-like carbons that exhibit XRD patterns with several diffraction peaks, very high surface area (approaching 4000 m2 g−1) and narrow micropore size distribution offers clear advantages over traditional activated carbons. The main benefit of microporous ZTCs is the high proportion of sharply distributed microporosity and near absence of mesopores, which contrasts with the high-surface-area activated carbons that tend to exhibit broad pore size distribution in the micropore and mesopore range. Zeolite templated carbons have so far shown great promise as gas storage materials, especially for hydrogen, where they already outperform most carbons and other porous materials. Studies have also shown that zeolite-templated carbons may be used as electrode materials for electrochemical capacitor applications. In future it would be interesting to explore the use of other microporous frameworks such as metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) as templates for carbons. More direct methods, such as soft templating, for synthesising highly ordered microporous carbon are also desirable.3. Mesoporous carbon materials
Since the first report on the synthesis of structurally well-ordered mesoporous silica via supramolecular self-assembly using long chain alkylammonium ions as surfactants,90,91 there has been rapid development in the synthesis of mesoporous silica with diverse structures and uniform pore sizes using various templates, including ionic surfactants, neutral amines and block copolymers, as structure-directing agents.90–94 The use of mesoporous silicas as solid templates to nanocast porous carbons is a natural development of the fast expanding field of mesoporous materials. Due to the availability of diverse structures of ordered mesoporous silica, the nanocasting of ordered mesoporous carbon from mesoporous silica is attractive and has developed rapidly over the past ten years. Different from microporous zeolites, mesoporous silicas usually have larger and controllable pore diameters, tunable wall thicknesses and a variety of pore geometries. Such excellent characteristics of ordered mesoporous silicas, including three-dimensional pore connectivity, clearly offer more flexibility in control of the structure, pore diameter, morphology and surface properties of templated mesoporous carbons.3.1 Conventional hard template synthesis strategy
3.1.1.1 Mesoporous silica MCM-48, SBA-15 and analogues as hard templates. Two Korean research groups, Ryoo and co-workers26 and Hyeon and colleagues5 were the first to independently report the synthesis of ordered mesoporous carbon using cubic mesoporous silica MCM-48 (Ia3d) as a hard template in 1999. Ryoo's group impregnated the pores of MCM-48 with sucrose solution containing sulfuric acid, followed by polymerisation, carbonisation and removal of the silica framework to obtain mesoporous carbon designated as CMK-1.26 As shown in Fig. 8, the XRD pattern of the carbon–silica composite was different from that of the template MCM-48 due to lattice contraction. The pore structure undergoes a systematic transformation to a new ordered cubic I4132 structure after removal of the silica template, as indicated in Fig. 8. The structural change is related to the disconnected nature of the two interwoven parts of the CMK-1 framework and involves their mutual displacement to create some contacts between them.95,96 The resulting CMK-1 mesoporous carbon has a high surface area and pore volume due to the presence of microporosity. The micropores are formed during the pyrolysis of the carbon precursor prior to removal of the silica framework, while mesopores of diameter 3 nm (Fig. 8) are formed after removal of the template.95
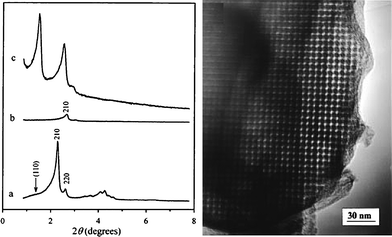 | ||
| Fig. 8 Left: Changes in powder XRD patterns during the synthesis of mesoporous carbon CMK-1 using mesoporous silica MCM-48 as the template: (a) MCM-48, (b) MCM-48/carbon composite after carbonisation within the MCM-48 pores, and (c) CMK-1 obtained by removing the silica from the composite. Right: TEM image of the ordered mesoporous carbon CMK-1. Reproduced with permission from ref. 26. | ||
On the other hand, Hyeon and co-workers used aluminosilica MCM-48 as the template and phenol resin as the carbon precursor to produce a mesoporous carbon named as SNU-1.5 The surface-implanted Al species acted as acid sites to catalyse the polymerisation of phenol and formaldehyde in the pores of the aluminosilica MCM-48. The mesoporous carbon SNU-1 was not a true negative replica of the MCM-48 template and had regular three-dimensionally interconnected 2 nm pores. The SNU-1 carbon exhibited excellent performance as an electrochemical double-layer capacitor.5
Over the past decade, mesoporous silicas including MCM-48,5 SBA-15,20,32,35,36 HMS,97 and MSU-H98 have been widely explored as templates for the preparation of ordered mesoporous carbon materials. Unlike the mesoporous carbon templated from MCM-48, the carbons templated from SBA-15, HMS and MSU-H are real inverse replicas of the silica template.5,26,32,97,98 Interestingly, the successful preparation of mesoporous carbon SNU-297 from mesoporous silica HMS and CMK-332 from mesoporous silica SBA-15 shed some light on the understanding of the structure of HMS and SBA-15 silicas. Originally, the HMS silica92 was proposed to be MCM-41-like with a hexagonal array of pore channels, but it turned out to possess a wormhole-like three-dimensionally interconnected pore structure.97 SBA-15 was initially thought to have a hexagonal tubular pore structure,94 similar to mesoporous silica MCM-41. However, later studies found that micropores or small mesopores exist between the primary cylindrical pore channels of SBA-15.99–101 The successful synthesis of mesoporous carbon CMK-3 confirmed the existence of such complementary micropores connecting the hexagonally packed mesopores in SBA-15 silica.32
Interestingly, depending on the degree of pore filling of the carbon precursor into the hexagonal (P6mm symmetry) pore system of mesoporous silica SBA-15, mesoporous carbons with different structures can be obtained. If the pore system of the SBA-15 is completely filled with carbon precursor, an ordered mesoporous carbon CMK-3 with P6mm symmetry, in which parallel carbon fibers are interconnected through thin carbon spacers, is generated.32 The pore structure of CMK-3 carbon is an inverse replica of SBA-15 silica. However, if the pore system of SBA-15 is partially coated by a carbon precursor, a surface-templated mesoporous carbon, named CMK-5, with an array of hollow carbon tubes, as shown by the TEM image in Fig. 9, is obtained.102 Moreover, due to the fact that the tubular structure exhibits both inner and outer surfaces, CMK-5 can have very high surface areas and large pore volumes, thus making it a potentially useful material for adsorption and catalyst-support applications. Partial coating of the carbon precursor on the surface of SBA-15 is considered crucial for the successful synthesis of CMK-5. Removal of the silica template then results in two different types of pores in the CMK-5 matrix. One type of pore is generated in the inner part of the channels that are not filled with carbon precursor. The other type of pore is obtained from the spaces where the silica walls of the SBA-15 template existed. Since there are two different mechanisms for pore generation, it should be possible to control the properties of the two pore systems independently. Schüth and co-workers explored the possibility of controlling these two pore system using aluminosilica SBA-15 as the template, and obtained ordered mesoporous carbon, (designated as NCC-1) with bimodal pore system and high pore volume.103 It was found that the crucial factors for the synthesis of such material are a synthesis temperature of 140 °C for the template SBA-15, a relatively low concentration of the carbon precursor furfuryl alcohol (25 vol%) and a carbonisation temperature higher than 750 °C. Several groups have reported other synthesis strategies to CMK-5 mesoporous carbon, such as controlling the polymerisation temperature and time,36,104–107 introducing the carbon precursor by catalytic CVD,108 and varying the concentration of furfuryl alcohol.109
 | ||
| Fig. 9 (a) TEM image of CMK-5 viewed along the direction of the ordered mesoporous carbon and the corresponding Fourier diffractogram, and (b) schematic model for the structure of CMK-5. Reproduced with permission from ref. 102. | ||
To synthesise bimodal porous carbons, a method was reported that combined nanocasting and imprinting strategies, wherein one pore system was generated by imprinting using colloidal particles and another mesopore system was nanocast from the mesoporous silica.110,111 The templating route allows the two pore systems to be well connected. The method allows one to tailor the bimodal distribution of mesopores by selecting silica colloids of desired size and SBA-15 particles of suitable pore wall thickness. Furthermore, the fraction of both types of pores can be controlled by adjusting the SBA-15:colloidal silica ratio.
Comparing mesoporous carbon materials templated from MCM-48 with those from SBA-15, it is obvious that the pore diameter for CMK-1 templated from MCM-48 is difficult to adjust since the wall thickness for MCM-48 is not easy to tune. However, the pore size for CMK-3 can be controlled by changing the synthesis conditions of the SBA-15 template. Using the triblock copolymer P123 as a structure-directing agent, Ryoo et al. and Zhao et al. synthesised large pore (3 to 10 nm) mesoporous silica materials with cubic Ia3d structures,112–115 from which large pore carbons were replicated. Ryoo and co-workers, used large pore cubic Ia3d KIT-6 mesoporous silica as a template to synthesise both rod-type and tube-type mesoporous carbon replicas,112,113 by controlling the polymerisation of furfuryl alcohol inside the silica pores. Zhao and co-workers used large pore three-dimensional bicontinuous cubic Ia3d FDU-5 mesoporous silica (prepared by solvent evaporation using the copolymer P123 as the template and the organosiloxane (3-mercaptopropyl)trimethoxysilane and trimethylbenzene as modifiers,114 to nanocast both rod-like and tube-like mesoporous carbons with bicontinuous cubic Ia3d symmetry.115,116 In contrast to CMK-1, carbon materials synthesised using KIT-6 and FDU-5 as templates show the same symmetry as the parent silica materials. The presence of porous bridges between the channel-like enantiomeric systems of these cubic silicas is responsible for structure retention in the replicated carbons, in a manner similar to that of CMK-3 where interconnecting micropores/small mesopores within the hexagonal cylindrical pores of SBA-15 silica are responsible for structure retention.32 Furthermore, in contrast to MCM-48, structure retention in the FDU-5 templated carbon arises from the formation of a rigid carbon framework that prevents symmetry change due to the larger pore size of FDU-5 compared to MCM-48.114
3.1.1.2 Other ordered mesoporous silicas as hard templates. Other mesoporous silicas including MCM-41, HMS, MSU-1, MSU-H, SBA-1, SBA-7, SBA-12, and SBA-16 have been explored as hard templates to fabricate mesoporous carbons. Mesoporous silica MCM-41, which has hexagonally ordered cylindrical one-dimensional pores,90,91 is unsuitable as a template since it yields disordered high-surface-area microporous carbon.48,97 This is due to the absence of complementary micropores within the MCM-41 silica walls. However, Zhao and co-workers prepared self-supported ordered ultrathin carbon nanowire arrays by employing MCM-41 silica as a template.117 The carbon nanowire arrays exhibit surface areas of up to 1400 m2 g−1, pore volumes of 1.1 cm3 g−1 and a uniform mesopore size of ca. 2.2 nm.
Mesoporous carbon templated by HMS (named as SUN-2) displays a bimodal pore system centered at 2.0 and 0.6 nm, which indirectly confirms that HMS silica possesses a wormlike pore structure rather than the originally proposed MCM-41-like hexagonal one-dimensional channel structure.97 Fuertes and co-workers found that the pore size and wall thickness of mesoporous HMS were tunable via changing the synthesis conditions,118 and using variously prepared HMS silicas as templates prepared carbons with large pore volumes of up to 3.5 cm3 g−1, high surface areas of up to 2300 m2 g−1, and narrow pore size distributions in the range 2–10 nm. They also adopted the same idea to tailor the pore size of mesoporous carbon by using MSU-1 silica as a template.37 MSU-H, a mesoporous silica with structure similar to SBA-15, which is synthesised under near-neutral conditions using sodium silicates as the silica source,119 is a cost-effective template for the synthesis of ordered mesoporous carbons.98
Mesoporous silica SBA-1 (which has a Pm3n cubic structure with two different kinds of mesoporous cages) was also used as a template for carbon formation.48 The resultant carbon, named as CMK-2, exhibited a low degree of structural ordering, which manifested itself by the presence of only one peak in the XRD pattern. The poor structural ordering of the CMK-2 carbon was explained by the small apertures in the cage-like mesoporous silica SBA-1 template, which make it difficult for the carbon precursor to fill the cages and to form rigid carbon bridges between carbon nanoparticles prepared in the silica cages.48 Li and co-workers employed SBA-1 and SBA-7 mesoporous silica as templates and sucrose as the carbon source to synthesise mesoporous carbon.120 They found that the pore size of cage-like mesoporous silica could be expanded under refluxing in acid solution without any addition of organic co-solvent, and that the obtained mesoporous silicas SBA-1 and SBA-7 could then be used as templates for the synthesis of mesoporous carbons with cubic Pm3n and three-dimensional hexagonal P63/mmc mesostructures.
Xia and Mokaya explored the synthesis of porous carbon materials via CVD using various mesoporous silicas including SBA-12, SBA-15, MCM-48, MCM-41 and HMS as templates, and acetonitrile as the carbon precursor.121 They achieved structural replication and high-surface-area N-doped mesoporous carbons from SBA-12, MCM-48, and SBA-15 silica templates. The N-doped carbons exhibited both well ordered mesoporosity and high levels of graphitic character depending on the CVD temperature and the nature of the silica template. It was found that higher CVD temperatures (>900 °C) generated high levels of graphitic character but compromised the mesostructural ordering of the carbon materials. The mesostructural ordering of the carbon materials and replication of pore channel ordering from the silica template was found to be dependent on the nature of the mesoporous silica used as the solid template.120
Ryoo and co-workers synthesised ordered mesoporous carbon using sucrose, furfuryl alcohol and acenaphthene as the carbon source and mesoporous silica SBA-16 with a cubic Im3m structure as the template.122 They found that furfuryl alcohol and acenaphthene were more suitable carbon precursors than sucrose for the formation of rigidly interconnected carbon bridges through narrow apertures of the cage-like silica SBA-16 mesostructure. Therefore the cubic Im3m structure was retained in the resulting mesoporous carbon product when fururyl alcohol was used. Guo et al. also synthesised mesostructured carbon materials with a cubic Im3m symmetry using mesoporous silica SBA-16 as a template.123 They found that the manner of template removal for the SBA-16 silica prior to use as a template played an important role in the subsequent carbon mesostructure templating process; solvent-extracted SBA-16 produced well-ordered mesostructured carbon while calcined SBA-16 gave poorly ordered carbons.
Jaroniec's group synthesised mesoporous carbons by using spherical silica particles as template and mesophase pitch or acrylonitrile as the carbon precursor.128 They also developed a colloid imprinting technique to synthesise mesoporous carbons17 by using commercially available colloidal silica as a template to generate mesoporous carbons whose uniform pore size was determined by the size of the colloidal silica spheres. The key to this method is the incorporation of spherical silica colloids into mesophase pitch particles.17,18,129 The colloid imprinted carbon can be further graphitised at high temperatures with significant retention of textural properties but with a reduction in pore size from 24 to 16 nm. Recently, Jaroniec's group has synthesised mesoporous carbons with extremely large pore volumes of up to 6 cm3 g−1via formation of a thin carbon film on the pore walls of colloidal silica templates followed by template dissolution.130 The pore volume and pore size could be tailored by controlling the carbon film thickness and the size of silica colloids used. Carbons with bimodal distribution of uniform mesopores were also formed by co-imprinting of spherical silica colloids and hexagonally ordered mesoporous particles of SBA-15 into mesophase pitch particles.110
Making use of constrained polymerisation of divinylbenzene on surfactant-modified colloid silica, Jang et al. prepared carbon nanocapsules and mesocellular foams.131 Later, they reported that mesoporous carbons with highly uniform and tunable mesopores could be fabricated by one-step vapour deposition polymerisation using colloidal silica nanoparticles as templates and poly(acrylonitrile) as the carbon precursor.132 Lu and co-workers recently realised the synthesis of spherical mesoporous carbons via an aerosol-based, one-step approach using colloidal silica particles and/or silicate clusters as templates.133
Mesoporous carbons at the three length scales of micrometric (2–8 mm), sub-micrometric (0.2–0.5 mm) and nanometric (10–20 nm) dimensions have been successfully synthesised.20 The micrometric carbon shows a perfectly spherical morphology and a unimodal pore system made up of structural mesopores of 3 nm. The carbons synthesised at the sub-micrometric and nanometric length scales exhibit bimodal porosity made up of structural pores of 3 nm derived from the silica framework and textural pores corresponding to the interparticle voids. Using bimodal mesoporous silica composed of 30–40 nm sized nanoparticles with 3.5 nm sized three-dimensionally interconnected mesopores as templates, bimodal mesoporous carbon having 4 nm sized framework mesopores and approximately 30 nm sized textural pores was synthesised.134
Mesocellular carbon foam with uniform large mesopores has been produced by partially impregnating mesocellular aluminosilicate foam with phenol/formaldehyde.135 As indicated in Fig. 10, during the synthesis, the carbon precursor was impregnated into the complementary mesopores while the filling of the primary cellular space was avoided. Using mesocellular silica foam with a main cell diameter of 27 nm and a window size of 11 nm as the template, the resulting mesocellular carbon foam shows a primary cell diameter of 27 nm and window size of 14 nm. Mesocellular carbon foam composed of nanometre-sized primary particles, and 40 nm pores, has also been prepared using hydrothermally synthesised MSU-F silica as a template and poly(furfuryl alcohol) as a carbon source.136 On the other hand, Tatsumi's group synthesised mesocellular carbon foam with a main cell size of 24 nm and window size of 18 nm via two successive impregnation (of sucrose) and carbonisation steps.137 The obtained mesoporous carbon had closed hollow spherical pores, while the carbon obtained by single step impregnation of sucrose had open mesocellular pores.
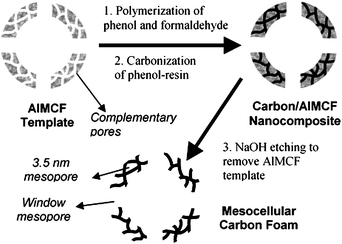 | ||
| Fig. 10 Schematic illustration of the synthesis of a mesocellular carbon foam. Reproduced with permission from ref. 135. | ||
3.2 Cost-effective strategies for the synthesis of mesoporous carbons
The disadvantage of traditional hard template methods for the synthesis of porous carbon is that they usually involve several steps starting with the preparation of a mesoporous silica/surfactant mesophase, followed by calcination to remove the surfactant (to generate the mesoporous silica hard template), introduction of carbon precursor into the mesoporous silica, carbonisation of the carbon precursor and finally silica etching (i.e., washing with hydrofluoric acid or NaOH) to generate the mesoporous carbon.26,32 Therefore, recently there have been some attempts to prepare mesoporous carbon via more direct methods involving fewer steps.Obviously, making use of as-synthesised mesoporous silica/surfactant mesophases as starting materials to synthesise mesoporous carbons is a cost-effective strategy since it needs fewer synthesis steps, and in particular does not require the mesophases to be calcined prior to use as template. The use of silica/surfactant mesophases as templates has been demonstrated by several research groups.137–140 Yu and co-workers138 prepared mesoporous carbon using an as-synthesised MCM-48 silica/surfactant mesophase as the template, followed by introduction of a carbon precursor (divinylbenzene), carbonisation and removal of the silica. On the other hand, Hyeon and co-workers139,140 reported the synthesis of mesoporous carbon by the carbonisation of composites containing silica, the triblock copolymer P123 and phenol resin, followed by removal of silica. The synthesis was achieved by treating the as-synthesised silica/triblock copolymer nanocomposite with sulfuric acid to crosslink the triblock copolymers followed by carbonisation.139 Pinnavaia and co-workers synthesised carbon nanotubes using P123 surfactant inside mesoporous silica.141
Other cost-effective strategies for synthesising mesoporous carbon involve even more direct synthesis procedures. For example, Kyotani and co-workers prepared mesoporous carbon via copolymerisation of TEOS and furfuryl alcohol.142 It was observed that the furfuryl alcohol/TEOS ratio significantly affected the mesopore size of the resulting carbon. Using a similar approach, Moriguchi et al. reported the direct synthesis of mesoporous carbon by in situ polymerisation of divinylbenzene in the hydrophobic phase of a hexagonally arrayed micelle/silicate nanocomposite followed by carbonisation and silica etching.143 The resulting carbon material had poor structural ordering with only wormhole-like mesopores of diameter 2 nm. The synthesis of mesoporous carbon via one-step vapour deposition polymerisation using colloidal silica particles as template and poly(acrylonitrile) as the carbon precursor has recently been reported by Jang and co-workers.144 Sayari and co-workers also presented a one-step in situ polymerisation route to nanoporous carbon via carbonisation of a cyclodextrin-templated silica mesophase, followed by removal of silica.145 Lu and co-workers have used a direct one-step aerosol process to prepare mesoporous carbon from sucrose solutions containing colloidal silica particles.146 Mesoporous carbons have also been fabricated via direct carbonisation of organic–organic nanocomposites comprising a thermosetting polymer and a thermally decomposable surfactant.147
The use of ordered mesoporous organosilicas as starting materials to synthesise mesoporous carbons presents a particularly interesting route since no extra carbon precursors are need. The direct synthesis of mesoporous carbon, with an average pore size of 2.5 nm, by carbonisation of phenyl-bridged mesoporous organosilica/surfactant mesophases followed by silica removal was reported by Lu et al.148 Mokaya's group also reported the direct preparation of nanostructured carbon materials from mesoporous ethyl-bridged organosilica/surfactant mesophases.149 By varying the pyrolysis temperatures, the textural properties of the resulting carbon are tunable, but the particle morphology of the organosilica is retained. Furthermore, mesoporous silica/carbon composites, mesoporous silica and silicon carbide materials can also be prepared from mesoporous ethyl-bridged organosilica/surfactant mesophases depending on the synthesis conditions.149
3.3 Soft-template synthesis strategy for ordered mesoporous carbons
It is noteworthy that, more recently, significant progress has been achieved on the direct synthesis of ordered mesoporous carbon materials by self-assembly of copolymer molecular arrays and carbon precursors. This opens a new way for the preparation of ordered mesoporous carbon materials with fewer synthesis steps. The self-assembly of organic–organic species via soft templating represents a breakthrough allowing the efficient synthesis of mesoporous polymers and mesoporous carbons with controlled pore structures. Dai and co-workers first reported the preparation of highly ordered and well-oriented mesoporous carbon thin films through the carbonisation of a nanostructured resorcinol–formaldehyde resin and self-assembled block copolymer poly(styrene)-block-poly(4-vinylpyridine) nanocomposite.150 The resulting mesoporous carbon thin films possess oriented cylindrical pores perpendicular to the substrate with dimensions of ca. 35 nm. They also prepared ordered mesoporous carbon structures in the form of monoliths, fibers, sheets, and films via self-assembly of triblock copolymer (Pluronic F127) with a mixture of phloroglucinol and formaldehyde.151 It is believed that hydrogen bonding between soft templates and carbon precursors is the driving force for successful organic–organic self-assembly. Similarly, Tanaka et al. reported the synthesis of ordered mesoporous carbon films with ordered hexagonal structure via direct carbonisation of organic–organic nanocomposites using resorcinol/formaldehyde and triethylorthoacetate as the carbon co-precursor and triblock copolymer Pluronic F127 as the template.147 Although there was no evidence to support triethylorthoacetate contributing to the carbon content of final carbon material, triethylorthoacetate was found to increase the periodicity of the porous carbon films.Zhao and co-workers have made major advances in the soft template synthesis of mesoporous carbons. They reported the self-assembly of the copolymer (PEO-PPO-PEO) templates and resol mixtures and successful removal of the templates including F127, F108 and P123 to produce mesoporous polymer and carbon materials.152–155Fig. 11 illustrates the five-step synthesis procedure adopted by Zhao's group. Low-molecular-weight phenolic resol is first mixed with PEO-PPO-PEO triblock copolymer in an ethanolic solvent, followed by the evaporation of the solvent which induces the self-assembly of the copolymer into an ordered structure. Driven by the hydrogen-bonding interaction between the PEO block and phenolic resol, an ordered mesostructure of the phenolic resol/block copolymer composite is formed. Curing of the resol at 100 °C solidifies the polymeric framework. Because of the difference in chemical and thermal stability between the resin and the triblock copolymer, the template can be removed either by calcination at 350–450 °C or by extraction with 48 wt% sulfuric acid solution, leaving the Bakelite framework with ordered aligned voids. Heating at a high temperature (above 600 °C) under an inert atmosphere transforms the polymeric framework to carbon mesostructures. The increase in both the resol/triblock copolymer ratio and the PEO content in the triblock copolymers results in mesophase transformation from lamellar, bicontinuous Ia3d, columnar p6mm to globular Im3m mesophases. The carbon mesostructures are highly stable and can be retained at temperatures as high as 1400 °C under a nitrogen atmosphere.153
 | ||
| Fig. 11 Scheme for the preparation of ordered mesoporous polymer resins and carbon frameworks. Reproduced with permission from ref. 154. | ||
Zhao and co-workers further explored other PEO-containing block copolymers as template to produce self-assembled carbon mesostructures.156 They found that high-molecular-weight PEO-PS block copolymers (PEO125-PS230) and low-molecular-weight phenolic resins can assemble into an ordered face-centered-cubic Fm3m mesophase. The pore size depends on the length of the hydrophobic PS blocks. A selective swelling of the PEO phase with phenolic resols can also be achieved in the reversed triblock copolymer PPO-PEO-PPO self-assembled structure.155 Two outer PPO blocks in a chain participate in two different micelles or aggregates, forming interconnected micelles. At certain molar ratios, the mixture of the reversed triblock copolymer PPO-PEO-PPO with large PEO weight fraction of 45% and phenolic resol self-assembles into face-centered-cubic packed spheres of PPO blocks with Fd3m symmetry in the PEO/phenolic resin matrix. A slight decrease in the phenolic resol/triblock copolymer ratio leads to the formation of a 2D hexagonal mesostructure. Interestingly, highly ordered mesoporous polymer and carbon frameworks with the Fd3m symmetry exhibit a bimodal pore-size distribution centered at 3.2–4.0 nm and 5.4–6.9 nm, respectively.
3.4 Ordered mesoporous carbons with graphitic pore walls
Despite their excellent structural ordering and narrow pore size distribution, the framework of most reported mesoporous carbons is generally not graphitic, i.e. the carbon framework is amorphous. For some applications, mesoporous carbons with graphitic pore walls are desirable. Mesoporous carbon (designated as CMK-3G) with a graphitic framework was prepared by Ryoo and co-workers viain situ conversion of aromatic compounds (including acenaphthene, acenaphthylene, indene, indane and naphthalene) to mesophase pitch inside mesoporous silica templates.157 The XRD pattern of CMK-3G was characterised by peaks at 2θ of 26, 45, 53 and 78°, which correspond to the (002), (101), (004) and (110) diffractions of graphitic carbon. TEM data indicated that the carbon framework consisted of discoid graphene sheets which are self-aligned perpendicular to the template walls thus giving the CMK-3G material improved thermal and mechanical stability. Pinnavaia and co-workers synthesised graphitic mesoporous carbon with high electrical conductivity using MSU-H silica as the template and various aromatic hydrocarbons as carbon precursors in the presence of a catalyst.158 They found that carbon materials obtained from pyrene and naphthalene precursors were more graphitic, whereas those from benzene exhibited the highest degree of conductivity. The methods reported by Ryoo157 and Pinnavaia158 required catalysts to convert the carbon precursor to graphitic mesoporous carbon.Fuertes and co-workers synthesised graphitic mesoporous carbons by liquid impregnation of poly(vinyl chloride) followed by carbonisation.159 Further heat treatment of the graphitisable carbon at high temperatures (up to 2300 °C) generated graphitised porous carbon with a relatively high surface area and porosity made up of mesopores in the 2–15 nm range. Later, they also produced mesoporous graphitic carbon from FeCl3-impregnated pyrrole at a lower temperature of 900 °C.160 FeCl3 acted not only as an oxidant for the polymerisation of pyrrole, but also as a catalyst for the formation of graphitic structure during the carbonisation step. Using mesophase pitch as the carbon precursor, Zhao's group prepared ordered graphitic mesoporous carbon with two-dimensional hexagonal P6mm symmetry or three-dimensional bicontinuous cubic Ia3d structure via a melting–impregnation method.161
The methods mentioned above for the preparation of ordered graphitic mesoporous carbon involve the use of liquid impregnation strategies, in which repeated infiltration and polymerisation, followed by carbonisation procedures are time consuming. CVD offers obvious advantages over liquid impregnation, such as time saving, a high degree of pore filling, enabling the formation of dense pore walls and avoids the formation of undesired microporosity. Xia and Mokaya have reported the synthesis of graphitic mesoporous carbon materials via a simple noncatalytic CVD method in which mesoporous SBA-15 is used as the solid template and styrene or acetonitrile as the carbon precursor.44 The degree of graphitisation is dependent on the CVD temperature with higher temperatures resulting in higher levels of graphitisation. Compared with styrene, the use of acetonitrile as the carbon precursor generates more highly graphitised carbon.44 The CVD method can be generalised for the preparation of various mesoporous graphitic carbon materials using mesoporous silica templates including SBA-12, SBA-15, MCM-48, HMS, and MCM-41.121 Xia, Yang and Mokaya also used CVD with acetonitrile as the precursor to prepare graphitic mesoporous carbon materials with diverse morphologies, such as sphere, hollow sphere, rod and nanotube.33,34,162,163 In a similar approach, Su et al. employed CVD, with benzene as the carbon source, to fabricate graphitic mesoporous carbon materials.164
3.5 Mesopore size control
The pore size of mesoporous carbon is of importance with respect to practical applications. When mesoporous carbon is synthesised via soft-templating methods that involve the self-assembly of organic–organic species, the pore size can be influenced by synthesis conditions, including surfactant type and concentration, and synthesis temperature. For example, Zhao and co-workers observed that the pore size of mesoporous carbon derived from soft-templated mesoporous polymer composites decreased from 7.4 to 5.9 nm when the pyrolysis temperature increased from 400 to 800 °C.153 However, it is more difficult to control the pore size of hard-templated mesoporous carbons. This is because control of pore size depends on changes in the wall thickness of the mesoporous silica templates.Ryoo and co-workers were the first to report pore size control in ordered mesoporous carbons by controlling the pore wall thickness of the silica template.165 By varying the ratio of two templates, cetyltrimethylammonium bromide and poly(oxyethylene hexadecylether)-type, in the synthesis of mesoporous silica, the wall thickness in the mesoporous silica changed by between 1.4–2.2 nm, which resulted in mesoporous carbons with pore size in the range 2.2–3.3 nm. Mokaya and co-workers have reported control of pore size in structurally well-ordered mesoporous carbons prepared via CVD nanocasting using SBA-15 rods as hard templates.163 The pore size was tunable from 2.0 to 4.3 nm by varying the synthesis temperature of the SBA-15 templates. Fuertes and co-workers reported the synthesis of mesoporous carbons with tailorable pore size using SBA-15, MSU-1 and HMS silica as a template.36,37,118 The pore size of the mesoporous carbon could be tuned continuously between 2 and 10 nm, by varying the synthesis temperature at which the MSU-1 or HMS templates were prepared. Moreover, by controlling the way the mesopores were filled by the carbon precursor, mesoporous carbon with unimodal or bimodal pore size distributions could be generated. As mentioned earlier, based on the two different mechanisms for the pore generation of CMK-5, Schüth and co-workers demonstrated that the intra-nanotube pore system can be tuned independently of the inter-nanotube pore system.103 More recently, Kim and co-workers reported the synthesis of ordered mesoporous carbons with controllable mesopore sizes in the 3 to 10 nm range, using boric acid as a pore expanding agent.166 They used a procedure similar to that reported by Ryoo et al.,26 except that boric acid was added along with sucrose as the carbon precursor. A phase separation of boron species during the carbonisation process was proposed as the genesis of the pore expansion.
3.6 Morphology control
The morphology of mesoporous carbons is another important factor with respect to their practical applications. Various macroscopic morphologies are required, for example, films (in sensor, separation and optics applications), uniformly sized spheres (in chromatography) or transparent monoliths (in electrodes). Using suitable synthesis strategies, it is possible to control the external shape of the templated mesoporous carbon materials to generate powders, films and membranes, spheres, hollow spheres, rods, fibers, nanowires, nanotubes and monoliths.3.6.2.1 Rods and spheres. Zhao and co-workers used mesoporous silica SBA-15 with rod-like morphology to prepare ordered mesoporous carbon rods.173 Sphere-shaped mesoporous carbon materials with controlled particle diameters ranging from 10 nm to 10 μm were prepared by Fuertes and co-workers using mesoporous silica spheres.20 Xia and Mokaya also synthesised spherical mesoporous carbon using spherical mesoporous silica SBA-15 as templates via a CVD route.33,34 Later, they synthesised mesoporous carbon nanorods via a CVD method using SBA-15 rod as templates (see Fig. 12). When rod-like mesoporous silica template synthesised at temperatures lower than 70 °C were used, mesoporous hollow nanotubules were obtained rather than solid core rods.163
 | ||
| Fig. 12 SEM images of mesoporous carbon nanocast via CVD using mesoporous silica SBA-15 rods as template; (a) an SBA-15 template synthesised at 40 °C and (b) an SBA-15 template synthesised at 100 °C.163 | ||
3.6.2.2 Hollow spheres. Porous carbon materials in the form of hollow spheres may find application in catalysis, controlled delivery, sensing and storage owing to their large volume and low density.174 Making use of hollow spherical mesoporous aluminosilicate as the template, Li et al. found that a bicontinuous mesostructure was faithfully and directly replicated to hollow spherical mesoporous carbon via a simple incipient-wetness impregnation technique.175 Depending on the synthesis conditions, hollow spherical carbon can also be obtained from mesoporous silica spheres. For example, carbon capsules with hollow core/mesoporous shell have been obtained using solid core mesoporous silica spheres as templates.176 The diameter of the hollow core and the mesoporous shell thickness were controlled by using appropriate solid core/mesoporous shell silica sphere templates.176 As shown in Fig. 13, Xia and Mokaya have successfully prepared well-ordered mesoporous carbon hollow spheres via a simple CVD method using SBA-15 as the template.33 The pyrolysis/carbonisation temperature was found to be very important and needed to be at least 950 °C for the successful formation of hollow spherical carbons. CVD temperatures lower than 950 °C resulted in the formation of mesoporous carbon with solid spheres. Xia and Mokaya also reported a synthesis route that utilised spherical solid core mesoporous silica SBA-15 as the template and optimised the morphology of the resulting mesoporous carbon exclusively toward hollow spheres.34 By changing the synthesis temperature of the SBA-15 template, hollow spherical mesoporous carbon with a pore size from 2 to 5 nm can be produced.162 Yu and co-workers also synthesised hollow core/mesoporous shell carbon using silicate-1 zeolite core/mesoporous silica shell structures as template.177
 | ||
| Fig. 13 Representative SEM (a) and (b) TEM images of hollow spheres of mesoporous carbon CMK-3.33,34 | ||
3.6.2.3 Films. Continuous mesoporous carbon thin films have been synthesised by Lu and co-workers through direct carbonisation of sucrose/silica nanocomposite films via a spin-coating technique and subsequent removal of the silica to create a mesoporous carbon network.178 The mesoporous carbon contains an average pore wall thickness of 2.0 nm and pore diameter of 2.4 nm. The continuous mesoporous carbon films display uniform-sized and interconnected pore channels, a high surface area of up to 2600 m2 g−1 and a high pore volume of 1.39 cm3 g−1. Recently, Lin et al. have synthesised ordered mesoporous thin film carbon with short channels vertical to the film by the replication of mesoporous silica SBA-15 film template, which has perpendicular channels obtained by using a ternary surfactant system (C16TMAB/SDS/P123) as the template.179 The mesoporous carbon film was deposited with a PtRu nanocatalyst and used as an anodic material in direct methanol fuel cells.180 The much enhanced methanol electrochemical oxidation activity in the PtRu/mesoporous carbon film was ascribed to efficient utilisation of the nanocatalysts within the short channels of the thin mesoporous carbon film.
3.6.2.4 Monoliths. Monolithic carbons are in some cases easier to handle than powdered materials. Direct shaping of monolithic mesoporous carbons during their preparation is often desirable, and may be achieved by using mesoporous silica monoliths as templates. Carbon monoliths with well-developed and accessible porosity have been produced using silica monoliths with hierarchical structure (containing macropores and mesopores) as templates and furfuryl alcohol or sucrose as carbon precursors.181–183 The generated carbons are positive replicas of the silica monolith at the micrometre level, and negative replicas on the nanometre scale. Interestingly, the pore system of the carbon monoliths can be varied to three- or four-modal porosity by varying the loading of the carbon precursor.181–183 Indeed, Shi et al. prepared carbon monoliths with bi- or tri-modal porosity from monolithic silica templates,184,185 while monolithic carbon may also be fabricated from monolithic colloidal silica.186
By integrating gel casting with chemical vapour deposition, Feng and co-workers reported the synthesis of hierarchical porous carbon monoliths with either hexagonal or cubic mesostructures starting from well-ordered mesoporous silica SBA-15 and KIT-6 powders.187 Powdery silica particles are first fused together to form silica monoliths by the gel-casting method. Furfuryl alcohol, at various concentrations in trimethylbenzene, was used as the carbon precursor. The use of a low concentration of furfuryl alcohol, together with a secondary loading of carbon via CVD, enabled greater control over the hierarchical porosity of the carbon monoliths. Monolithic mesoporous carbon with a bicontinuous cubic structure (Ia3d symmetry) prepared by using mesoporous silica monoliths as the hard template116 exhibits a uniform pore size of 4.6 nm and a surface area of 1530 m2 g−1, and is a promising electrode for electrochemical double-layer capacitors.
Xia and Mokaya have prepared ordered mesoporous carbon monoliths using mesoporous silica monoliths as template via CVD.188 The size and shape of the silica monolith is retained in the carbon monolith, making it mechanically robust. A relatively high level of mesostructural ordering with a low degree of graphitic character is observed in the CVD-generated carbon monolith. The mesoporous carbon monolith had considerable hydrogen uptake capacity of 3.4 wt% at 20 bar and −196 °C, which is comparable to that of powder forms of CMK-3 mesoporous carbon. Hu et al. have also synthesised carbon monoliths that possess both mesopores and macropores using meso/macroporous silica as a template and mesophase pitch as the carbon precursor.189 The bimodal carbon monoliths have a superior high-rate performance as an anode material in electrochemical lithium cells. It is believed that the monolith's high porosity (which provides ionic transport channels), higher graphitisation and enhanced electronic conductivity (ca. 0.1 S cm−1) are responsible for the high performance in lithium battery applications.189
3.6.2.5 Other morphologies. Mesoporous carbon materials with diverse morphology including monodisperse nanocubes and uniform nanospheres or tetrapods have been prepared by Stein and co-workers using block copolymers as the mesopore-directing agent and colloidal crystals as molds for the external shape of the particles.190 In a related approach, mesoporous carbon nanowires, fibers and nanotubes with high aspect ratios and low defect density may be fabricated using porous alumina membranes as confinement matrices.191–194 Such confined growth of mesostructures inside the voids of porous alumina membranes is an effective approach to make fibers or nanowires since the macroscopic morphology of porous alumina membranes is maintained in the resulting mesoporous nanowires or fibers. For example, Bein et al. have successfully prepared hierarchically oriented mesoporous carbon nanofilaments as replicas of silica mesostructures deposited inside the channels of anodic alumina membranes.193 Due to the confinement imposed by the channels of the alumina membranes, rather unusual mesophase structures showing “circular” or “columnar” mesoscopic channels were formed, and replicated as carbon analogues after removal of the templating hosts. Later, the same group fabricated well-aligned free-standing mesoporous carbon nanofiber arrays using the nonionic triblock copolymer F127 as a soft template and porous alumina membranes as hard templates via a confined self-assembly process.192 Hexagonally arranged circular mesochannels were located at the edge of the carbon nanofibers while well-ordered columnar oriented mesochannels were wrapped by circular mesochannels at the center of the nanofibers. Indeed the confinement synthesis of mesostructured porous materials has opened new opportunities for fabrication of mesoporous materials.195–197 The attraction of confinement synthesis is that it can generate templated mesoporous carbons with unusual morphology, whereby the mesoscale porosity is controlled via soft or hard templating while the particle morphology is determined by the shape of the confined matrix space.
4. Macroporous carbon materials
Spherical submicrometer-sized particles such as polystyrene and silica can self-organise to form colloidal crystals, known as opals, which are excellent templates for three-dimensional macroporous carbon materials with hollow or core/shell structures. The pore size of the macroporous carbons is tunable by changing the size of the spherical silica or polymer particles. To obtain macroporous carbon materials, colloid crystals are first formed via packing uniform spherical silicas into two- or three-dimensional arrays. Filling of carbon precursors into the interstitial space of the colloid crystals, carbonisation and finally removal of the spherical templates generate a carbon skeleton in the location of the former interstitial space and interconnected voids where the spheres were originally located. It is worth mentioning that before the infiltration of the carbon precursors, sintering is usually performed to create necks between the silica spheres, which provide interconnections between the spherical pores in the macroporous carbons.4.1. Silica colloidal crystals as hard templates
The synthesis of macroporous carbon materials was first realised by Zakhidov et al. in 1998.38 Macroporous carbon materials with inverse opal structures were obtained using silica opals as hard templates and phenol resin and/or propylene gas as the carbon precursor. The macroporous carbons had different structures depending on the synthesis conditions; diamond and glassy carbon inverse opals were formed via volume filling, while graphite inverse opals, comprising 4 nm-thick layers of graphite sheets tiled on spherical surfaces were produced by surface templating.37In principle, the morphology of macroporous carbon materials is largely dependent on the degree of void infiltration of the opal template. In order to maximise the filling of the interstitial voids with carbon precursors, liquid-phase carbon precursors such as phenolic resin and sucrose solution are usually used to achieve better replication.38,39,198–200 A variety of carbon precursors, including propylene gas, benzene and divinylbenzene can also be successfully utilised to make three-dimensional macroporous carbon materials using colloid crystals as hard templates.38,201,202 The resulting macroporous carbons can exhibit large pore volume and high surface area. For example, macroporous carbon synthesised using phenol resin as the carbon precursor had close-packed spherical pores with a diameter of 62 nm, a total pore volume of 1.68 cm3 g−1, and a surface area of 750 m2 g−1.203 However, it is now known that during the carbonisation and pyrolysis processing, the carbon phase can shrink remarkably. To achieve highly ordered carbon structures, it is therefore necessary to perform repeated impregnation cycles. Perpall et al. reported the synthesis of an inverse carbon opal using low-shrinkage bis-ortho-diynyl arene monomers as precursor and found that the carbon inverse opal structure retained the original dimensions of the template.204
The surface properties of the colloid spheres and the chemical nature of the carbon precursors are important parameters in carbon growth. Consequently, the morphology of macroporous carbon materials can be controlled by altering the surface properties of colloid spheres and choosing suitable carbon precursors. Usually, good adhesion of the carbon precursor to the colloid surface results in a surface-templating mechanism. For this reason, the position of carbon growth can be controlled via chemical modification of the colloid surface. Yu and co-workers reported the synthesis of three-dimensional ordered macroporous carbon materials with various morphologies by using Al-impregnated silica spheres as the template, which allowed control of the initiation sites of the acid-catalyzed carbonisation reaction of phenol and formaldehyde.39 An Al-grafted silica array results in surface coating, namely surface templating, as shown in Fig. 14a. In this case, polymerisation is initiated at the acidic sites on the Al-grafted silica surface. However, polymerisation is thought to occur everywhere to fill up the entire space between the particles when an acid catalyst is mixed with a carbon solution. Fig. 14b clearly shows that an ordered macroporous carbon framework was formed by the volume-templating mechanism with complete filling of all the voids in the silica spheres. Each of the spherical pores is interconnected though small channels. Yu and co-workers also reported the synthesis of ordered uniform porous carbon with pore size in the range of 10–1000 nm using crystalline silica spheres as templates via carbonisation of phenol with formaldehyde.198 The resulting porous carbons show a variety of porous structures, which can be easily controlled by tuning the size of colloid silica spheres.
 | ||
| Fig. 14 SEM images of macroporous carbon materials fabricated (a) by surface templating and (b) by volume templating using 250 nm silica spheres as the template. The inset shows a carbon–colloidal silica composite. Reproduced with permission from ref. 39. | ||
4.2 Polymer microspheres as templates
The use of polymer microspheres as templates has the advantage of eliminating the chemical dissolution step since the polymer template can simply wash away or be burnt. The use of polystyrene microspheres as templates for the synthesis of ordered macroporous carbons was realised by Baumann and co-workers.40 Following infiltration of hydrogel into the interstitial spaces between polystyrene colloid spheres, the polystyrene sphere template was removed by washing with toluene, after which heat treatment generated an ordered macroporous carbon with 100 nm cavities and 6 nm interconnections. Macroporous carbon doped with various metal nanoparticles could be prepared when a metal ion-doped hydrogel was used as the carbon source since metal ions were reduced to metal nanoparticles during the carbonisation step.Recently, Antonietti and co-workers reported a soft-template-based method for the synthesis of carbon with meso- and macroporosity in a one-step process, taking advantage of the phase separation (spinodal decomposition) of mesophase pitch, which acted as the carbon precursor, and a commercially available organic polymer polystyrene, which acted as a soft template.205 Due to the use of soluble mesophase pitch and polystyrene, it is possible to control the morphology of the resulting carbon materials to monoliths or films.205 Liang and Dai used a similar strategy to synthesise bimodal meso/macroporous carbon monoliths through a dual-phase separation process in which microphase separation occurred in the polymer phase and spinodal decomposition progressively developed through the crosslinking of the corresponding polymer mixture.206 The bimodal porous nature of the resulting carbon monoliths was derived from the dual-phase separation in which spinodal decomposition and microphase separation occurred simultaneously. The macropores evolved from the solvent-rich phase and the mesopores were generated after pyrolysis of the block copolymer templates.
4.3. Dual-template method
Chai et al. developed a dual-template method for the fabrication of macrostructurally patterned, highly ordered, three-dimensionally interconnected porous carbon with uniform mesopore walls.202 They used both monodisperse polystyrene spheres and silica particles as templates and divinylbenzene as carbon precursor. Both macroporosity and mesoporosity could be manipulated in the resulting carbon by controlling the sphere size of polystyrene and silica particles. The introduction of secondary mesopores into a three-dimensionally ordered macroporous carbon skeleton by templating with secondary silica nanoparticles led to improved electrochemical activity when the bi-modal carbon was employed as a catalyst support in a direct methanol fuel cell.202 Woo and co-workers also reported the synthesis of hierarchical three-dimensional ordered macroporous carbons with walls composed of hollow mesosized spheres via a dual-templating strategy.207 Macropores were created by using poly[styrene-co-(2-hydroxyethyl methacrylate)] polymeric colloids as hard templates and hollow mesosized spheres were templated by smaller silica colloids. Thus, two types of colloids of significantly different sizes were used to create a macrosized polymeric colloidal crystal with voids filled with mesosized smaller silica colloids. The large macropores were interconnected and three-dimensionally ordered, and the walls of the large macropores were composed of small hollow carbon spheres, as shown in Fig. 15. The macropore size and the hollow carbon sphere size could be controlled simply by choosing the size of polymer and silica particles, respectively. | ||
| Fig. 15 SEM images of porous carbons prepared using hard templates consisting of large polymer colloids (450 nm diameter) and small silica colloids. The particle size of silica colloids used as template for the small spherical pores is (a) 70–100 nm and (b) 40–50 nm.207 | ||
The use of dual-templating strategies to synthesise three-dimensional macroporous carbon materials has been explored by the groups of Zhao and Stein.208–210 In this case, the two templates play different roles: one as a hard template to control the macroscopic structures, and the other as a soft template for self-assembly of structurally ordered mesopores. Zhao and co-workers prepared hierarchically ordered macro/mesoporous carbons using monodispersed silica colloidal crystals as hard templates, amphiphilic triblock copolymer PEO-PPO-PEO as a soft template, and soluble resols as a carbon source.208 As shown in Fig. 16, the obtained porous carbons have a highly ordered face-centered-cubic macrostructure with tunable pore sizes of 230–430 nm and interconnected windows with a size of 30–65 nm. The rigid silica hard templates prevent shrinkage of the mesostructure during the thermosetting and carbonisation processes, resulting in large cell parameters of 18 nm and pore sizes of 11 nm. The bimodal porous carbon materials have large surface area of up to 760 m2 g−1, high pore volume of up to 1.25 cm3 g−1, and partially graphitised frameworks. With the increase in the silica sphere diameter, the surface areas and the window sizes increase.205
 | ||
| Fig. 16 (a) TEM images of ordered macro/mesoporous carbons with macropore arrays viewed along the (111) planes and (b) high-magnification TEM images, showing the carbon mesostructure. Reproduced with permission from ref. 208. | ||
Stein's group, on the other hand, have reported the synthesis of three-dimensionally ordered macro/mesoporous carbon monoliths using poly(methyl methacrylate) (PMMA) colloidal crystals as templates and a concentrated triconstituent precursor solution (a soluble phenolformaldehyde prepolymer, tetraethylorthosilicate, and the nonionic triblock copolymer F127) as the carbon precursor.210 The PMMA colloidal crystal template permits control over the external morphology of the carbon products. It is possible to produce either monoliths with hierarchical porosity (ordered macropores from PMMA spheres and large mesopores from F127) or cubic and spherical mesoporous nanoparticles. The specific morphology depends on the concentration of F127 and on the presence of 1,3,5-trimethyl benzene as an additive. Stein and co-workers also found that hierarchically ordered macroporous polymers and carbon monoliths with walls containing face-centered-cubic or 2D-hexagonal mesopores can be synthesised via a dual-templating technique using PMMA colloidal crystals and an amphiphilic triblock copolymer as templates.209 The mesostructures could be conveniently controlled by tuning the concentration of the copolymer. A two-step thermal curing strategy was adopted to achieve a highly cross-linked mesostructure composed of a phenolic resin and a block copolymer. This ensured the formation of robust mesopore walls containing phenolic resin that survived during the decomposition of the block copolymer template. The ordered mesoporous phenolic resin was subsequently transformed to mesoporous carbon after heat treatment at high temperature under an inert atmosphere. The growth of mesopores was significantly influenced by the confinement effect of the colloidal crystal template. Both spherical and cylindrical mesopores were aligned parallel to the surface of PMMA spheres, and therefore the obtained mesostructures exhibited apparent curvatures near the surface of macropore walls. The macroporous carbons produced via this method were mechanically more stable than hierarchically porous carbon monoliths synthesised by conventional nanocasting.
Three-dimensional macroporous carbon materials usually possess low or no crystallinity due to the relatively low carbonisation temperature, which is usually lower than 1000 °C. To increase the crystallinity of macroporous carbon to form graphitic carbon, high-temperature annealing is required. Recently, Jaroniec and colleagues reported the preparation of highly ordered graphitised mesoporous/macroporous carbons using commercial mesophase pitch as a carbon precursor and silica colloidal crystals as templates.211 The synthesis of the graphitised ordered nanoporous carbon was carried out by the incorporation of mesophase pitch dissolved in quinoline in the interstitial space of the silica templates under a static vacuum. After carbonisation and the removal of the silica template, the resulting carbon was further heated at high temperature of up to 2500 °C in argon atmosphere to generate highly graphitised carbon. The XRD patterns of the carbon material after the graphitisation at 2500 °C showed a sharp (002) peak with an interlayer spacing of 0.33 nm, and two further peaks, corresponding to graphitic (101) and (004) diffractions. After graphitisation, the carbon material possessed both mesopores and macropores of size in the range 40–100 nm that are interconnected, and exhibited relatively large graphite crystallites in the carbon pore walls.
5. Concluding remarks
Hard templating is a versatile method for the preparation of nanoscale porous carbon materials, and tremendous progress has been made in the past decade. Carbon materials with diverse pore systems, morphology and pore size have been realised via hard-templating routes. By using this approach, and depending on the structural characteristics of the host template, it is possible to synthesise structurally well-ordered nanoscale porous carbons with highly refined textural characteristics, uniform and tunable particle size, and a narrow pore size distribution with pore diameters varying from micropores to mesopores and macropores. Since there are plenty of hard templates with periodic porous structures, which are either commercially available (e.g. zeolites and clays) or generated via soft-template synthesis and/or sol–gel chemistry (e.g. mesoporous or macroporous silicas), the hard template route is likely to remain a versatile tool that will be further developed. However, the large-scale production of nanoscale templated porous carbons via the hard template route is inevitably hindered by its intrinsic limitations: the sacrificial use of hard templates and the use of toxic hydrofluoric acid (although NaOH may be used) to etch silicate templates.Soft templating of porous nanoscale carbons represents a new synthesis strategy which makes use of supramolecular assembly between carbon polymeric precursors and template molecules (e.g., surfactants or copolymers). Although the soft template route to ordered porous carbons is still in its early research stage, it definitely provides opportunities to overcome the limitations of hard templates and can provide nanostructured carbons in large quantities. Moreover, soft templating offers the chance to synthesise nanoscale porous carbon materials with particular structures which are difficult to realise via hard templates. The increasing demand for optimised nanoscale carbons in new technologies guarantees the continuing development of templating routes to carbons.
However, there are still plenty of challenges ahead. For example, unlike mesoporous carbons that can be prepared with various pore sizes via either hard or soft template methods, microporous carbons with tunable pore diameter and narrow pore size distribution remain a major challenge. This is because the wall thickness of zeolite templates is too rigid to adjust. Preparation of highly ordered (well defined pore diameter) and high surface area microporous carbons via direct routes is a challenge that will continue to attract attention due to the fact that such microporous carbons are of great importance for energy applications such as gas storage. Likely applications include storage of hydrogen or methane and sequestration of carbon dioxide, and use as electrode materials for fuel cells.
To date, block copolymers have dominated as soft templates for the synthesis of mesoporous carbons and only hydrogen bonding has been explored as the self-assembly driving force. It is desirable to investigate other driving forces and other surfactants or templates to synthesise porous carbon materials. In particular, the synthesis of microporous carbon via soft template strategies is of great interest. The further development of new hierarchical materials that possess not only mesopores and macropores, but also micropores will be desirable for new applications involving smaller molecules.
References
- Y. Mastai, S. Polarz and M. Antonietti, Adv. Funct. Mater., 2002, 12, 197–202 CrossRef CAS
.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS
- M. Kang, S. H. Yi, H. I. Lee, J. E. Yie and J. M. Kim, Chem. Commun., 2002, 1944–1945 RSC
- Z. Yang, Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 727–732 CrossRef CAS
- J. Lee, S. Yoon, T. Hyeon, S. M. Oh and K. B. Kim, Chem. Commun., 1999, 2177–2178 RSC
- B. Sakintuna and Y. Yueruem, Ind. Eng. Chem. Res., 2005, 44, 2893–2902 CrossRef CAS
- T. Kyotani, Carbon, 2000, 38, 269–286 CrossRef CAS
- H. C. Foley, Microporous Mater., 1995, 4, 407–433 CrossRef CAS
-
C. R. Bansal, J. B. Donnet and F. Stoeckli, Active carbon, Marcel Dekker, New York, 1988 Search PubMed
- K. Miura, J. Hayashi and K. Hashimoto, Carbon, 1991, 29, 653–660 CrossRef CAS
- H. Nakagawa, K. Watanabe, Y. Harada and K. Miura, Carbon, 1999, 37, 1455–1461 CrossRef CAS
- F. Cheng, J. Liang, J. Zhao, Z. Tao and J. Chen, Chem. Mater., 2008, 20, 1889–1895 CrossRef CAS
- A. Oya, S. Yoshida, J. Alcaniz-Monge and A. Linares-Solano, Carbon, 1995, 33, 1085–1090 CrossRef CAS
- N. R. Khalili, M. Campbell, G. Sandi and J. Golas, Carbon, 2000, 38, 1905–1915 CrossRef CAS
- J. Ozaki, N. Endo, W. Ohizumi, K. Igarashi, M. Nakahara, A. Oya, S. Yoshida and T. Iizuka, Carbon, 1997, 35, 1031–1033 CrossRef CAS
- H. Tamon, H. Ishizaka, T. Araki and M. Okazaki, Carbon, 1998, 36, 1257–1262 CrossRef CAS
- Z. Li and M. Jaroniec, J. Am. Chem. Soc., 2001, 123, 9208–9209 CrossRef CAS
- Z. Li and M. Jaroniec, J. Phys. Chem. B, 2004, 108, 824–826 CrossRef CAS
- Z. Li, M. Jaroniec, Y. Lee and L. R. Radovic, Chem. Commun., 2002, 1346–1347 RSC
- A. B. Fuertes, J. Mater. Chem., 2003, 13, 3085–3088 RSC
- T. Kyotani, Bull. Chem. Soc. Jpn., 2006, 79, 1322–1337 CrossRef CAS
- Y. Wan, Y. F. Shi and D. Y. Zhao, Chem. Mater., 2008, 20, 932–945 CrossRef CAS
- F. Schüth, Angew. Chem., Int. Ed., 2003, 42, 3604–3622 CrossRef
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef CAS
- J. H. Knox, B. Kaur and G. R. Millward, J. Chromatogr., A, 1986, 352, 3–25 CrossRef CAS
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS
- C. Vix-Guterl, S. Saadallah, L. Vidal, M. Reda, J. Parmentier and J. Patarin, J. Mater. Chem., 2003, 13, 2535–2539 RSC
- T. Kyotani, T. Nagai, S. Inoue and A. Tomita, Chem. Mater., 1997, 9, 609–615 CrossRef CAS
- T. Kyotani, Z. X. Ma and A. Tomita, Carbon, 2003, 41, 1451–1459 CrossRef CAS
- J. Rodriguez-Mirasol, T. Cordero, L. R. Radovic and J. J. Rodriguez, Chem. Mater., 1998, 10, 550–558 CrossRef CAS
- Z. X. Ma, T. Kyotani and A. Tomita, Chem. Commun., 2000, 2365–2366 RSC
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS
- Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 886–891 CrossRef CAS
- Y. Xia, Z. Yang and R. Mokaya, J. Phys. Chem. B, 2004, 108, 19293–19298 CrossRef CAS
- A. Lu, A. Kiefer, W. Schmidt and F. Schueth, Chem. Mater., 2004, 16, 100–103 CrossRef CAS
- A. B. Fuertes, Microporous Mesoporous Mater., 2004, 67, 273–281 CrossRef CAS
- S. Alvarez and A. B. Fuertes, Carbon, 2004, 42, 433–436 CrossRef CAS
- A. A. Zakhidov, R. H. Baughman, Z. Iqbal, C. X. Cui, I. Khayrullin, S. O. Dantas, I. Marti and V. G. Ralchenko, Science, 1998, 282, 897–901 CrossRef CAS
- J. S. Yu, S. Kang, S. B. Yoon and G. Chai, J. Am. Chem. Soc., 2002, 124, 9382–9383 CrossRef CAS
- T. F. Baumann and J. H. Satcher, Chem. Mater., 2003, 15, 3745–3747 CrossRef CAS
- M. Alvaro, P. Atienzar, J. L. Bourdelande and H. Garcia, Chem. Commun., 2002, 3004–3005 RSC
- C. J. Meyers, S. D. Shah, S. C. Patel, R. M. Sneeringer, C. A. Bessel, N. R. Dollahon, R. A. Leising and E. S. Takeuchi, J. Phys. Chem. B, 2001, 105, 2143–2152 CrossRef CAS
- F. B. Su, X. S. Zhao, L. Lu and Z. C. Zhou, Carbon, 2004, 42, 2821–2831 CrossRef CAS
- Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 1553–1558 CrossRef CAS
- Z. X. Yang, Y. D. Xia and R. Mokaya, Microporous Mesoporous Mater., 2005, 86, 69–80 CrossRef CAS
- P. X. Hou, H. Orikasa, T. Yamazaki, K. Matsuoka, A. Tomita, N. Setoyama, Y. Fukushima and T. Kyotani, Chem. Mater., 2005, 17, 5187–5193 CrossRef CAS
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS
- A. H. Lu and F. Schuth, Adv. Mater., 2006, 18, 1793–1805 CrossRef CAS
- A. Stein, Z. Y. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS
- F. B. Su, Z. C. Zhou, W. P. Guo, J. J. Liu, X. N. Tian and X. S. Zhao, Chem. Phys. Carbon, 2008, 30, 63–128 Search PubMed
- H. Yang and D. Zhao, J. Mater. Chem., 2005, 15, 1217–1231 RSC
- J. Lee, S. Han and T. Hyeon, J. Mater. Chem., 2004, 14, 478–486 RSC
- B. Sakintuna and Y. Yurum, Ind. Eng. Chem. Res., 2005, 44, 2893–2902 CrossRef CAS
- G. Frey and A. K. Cheetham, Science, 1999, 283, 1125–1126 CrossRef CAS
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS
- P. Enzel and T. Bein, Chem. Mater., 1992, 4, 819–824 CrossRef CAS
- T. J. Bandosz, J. Jagiello, K. Putyera and J. A. Schwarz, Chem. Mater., 1996, 8, 2023–2029 CrossRef CAS
- P. M. Barata-Rodrigues, T. J. Mays and G. D. Moggridge, Carbon, 2003, 41, 2231–2246 CrossRef CAS
- A. Garsuch and O. Klepel, Carbon, 2005, 43, 2330–2337 CrossRef CAS
- A. Garsuch, O. Klepel, R. R. Sattler, C. Berger, R. Glaser and J. Weitkamp, Carbon, 2006, 44, 593–596 CrossRef CAS
- H. Nishihara, Q. H. Yang, P. X. Hou, M. Unno, S. Yamauchi, R. Saito, J. I. Paredes, A. Martinez-Alonso, J. M. D. Tascon, Y. Sato, M. Terauchi and T. Kyotani, Carbon, 2009, 47, 1220–1230 CrossRef CAS
- S. A. Johnson, E. S. Brigham, P. J. Ollivier and T. E. Mallouk, Chem. Mater., 1997, 9, 2448–2458 CrossRef CAS
- Z. X. Ma, T. Kyotani, Z. Liu, O. Terasaki and A. Tomita, Chem. Mater., 2001, 13, 4413–4415 CrossRef CAS
- P. X. Hou, T. Yamazaki, H. Orikasa and T. Kyotani, Carbon, 2005, 43, 2624–2627 CrossRef CAS
- F. Su, J. Zeng, Y. Yu, L. Lu, J. Y. Lee and X. S. Zhao, Carbon, 2005, 43, 2366–2373 CrossRef CAS
- C. O. Ania, V. Khomenko, E. Raymundo-Pinero, J. B. Parra and F. Beguin, Adv. Funct. Mater., 2007, 17, 1828–1836 CrossRef CAS
- F. O. M. Gaslain, J. Parmentier, V. P. Valtchev and J. Patarin, Chem. Commun., 2006, 991–993 RSC
- S. Lei, J. I. Miyamoto, T. Ohba, H. Kanoh and K. Kaneko, J. Phys. Chem. C, 2007, 111, 2459–2464 CrossRef CAS
- P. Srinivasu, A. Vinu, N. Gokulakrishnan, S. Anandan, A. Asthana, T. Mori and K. Ariga, J. Nanosci. Nanotechnol., 2007, 7, 2913–2916 CrossRef CAS
- A. P. Wang, F. Y. Kang, Z. H. Huang and Z. C. Guo, New Carbon Mater., 2007, 22, 141–147 Search PubMed
- Z. X. Yang, Y. D. Xia, X. Z. Sun and R. Mokaya, J. Phys. Chem. B, 2006, 110, 18424–18431 CrossRef CAS
- Z. X. Yang, Y. D. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673–1679 CrossRef CAS
- A. Pacula and R. Mokaya, J. Phys. Chem. C, 2008, 112, 2764–2769 CrossRef CAS
- Y. Xia, G. S. Walker, D. M. Grant and R. Mokaya, J. Am. Chem. Soc., 2009, 131, 16493–16499 CrossRef CAS
- C. Portet, Z. Yang, Y. Korenblit, Y. Gogotsi, R. Mokaya and G. Yushin, J. Electrochem. Soc., 2009, 156, A1–A6 CrossRef CAS
- N. Sonobe, T. Kyotani and A. Tomita, Carbon, 1988, 26, 573–578 CAS
- N. Sonobe, T. Kyotani and A. Tomita, Carbon, 1990, 28, 483–488 CrossRef CAS
- N. Sonobe, T. Kyotani and A. Tomita, Carbon, 1991, 29, 61–67 CrossRef CAS
- T. Kyotani, H. Yamada, N. Sonobe and A. Tomita, Carbon, 1994, 32, 627–635 CrossRef CAS
- T. J. Bandosz, K. Putyera, J. Jagiello and J. A. Schwarz, Carbon, 1994, 32, 659–664 CrossRef CAS
- T. J. Bandosz, J. Jagiello, K. Putyera and J. A. Schwarz, Langmuir, 1995, 11, 3964–3969 CrossRef CAS
- T. Kyotani, T. Mori and A. Tomita, Chem. Mater., 1994, 6, 2138–2142 CrossRef CAS
- K. Putyera, T. J. Bandosz, J. Jagieo and J. A. Schwarz, Carbon, 1996, 34, 1559–1567 CrossRef CAS
- G. Sandí, P. Thiyagarajan, K. A. Carrado and R. E. Winans, Chem. Mater., 1999, 11, 235–240 CrossRef CAS
- G. Sandi, K. A. Carrado, R. E. Winans, J. R. Brenner and G. W. Zajac, Microporous Macroporous Mater., 1996, 431, 39–44 Search PubMed
- A. Pacuła and R. Mokaya, Microporous Mesoporous Mater., 2007, 106, 147–154 CrossRef CAS
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865–867 CrossRef CAS
- P. T. Tanev and T. J. Pinnavaia, Science, 1996, 271, 1267–1269 CrossRef CAS
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Frederickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS
- M. Kruk, M. Jaroniec, R. Ryoo and S. H. Joo, J. Phys. Chem. B, 2000, 104, 7960–7968 CrossRef CAS
- M. Kaneda, T. Tsubakiyama, A. Carlsson, Y. Sakamoto, T. Ohsuna, O. Terasaki, S. H. Joo and R. Ryoo, J. Phys. Chem. B, 2002, 106, 1256–1266 CrossRef CAS
- J. Lee, S. Yoon, S. M. Oh, C. Shin and T. Hyeon, Adv. Mater., 2000, 12, 359–362 CrossRef CAS
- S. S. Kim and T. J. Pinnavaia, Chem. Commun., 2001, 2418–2419 RSC
- R. Ryoo, C. H. Ko, M. Kruk, V. Antochshuk and M. Jaroniec, J. Phys. Chem. B, 2000, 104, 11465–11471 CrossRef CAS
- M. Impéror-Clerc, P. Davidson and A. Davidson, J. Am. Chem. Soc., 2000, 122, 11925–11933 CrossRef CAS
- M. Kruk, M. Jaroniec, C. H. Ko and R. Ryoo, Chem. Mater., 2000, 12, 1961–1968 CrossRef CAS
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS
- A. Lu, W. Schmidt, B. Spliethoff and F. Schueth, Adv. Mater., 2003, 15, 1602–1606 CrossRef CAS
- L. A. Solovyov, T. Kim, F. Kleitz, O. Terasaki and R. Ryoo, Chem. Mater., 2004, 16, 2274–2281 CrossRef CAS
- S. N. Che, K. Lund, T. Tatsumi, S. Iijima, S. H. Joo, R. Ryoo and O. Terasaki, Angew. Chem., Int. Ed., 2003, 42, 2182–2185 CrossRef CAS
- M. Kruk, M. Jaroniec, T. Kim and R. Ryoo, Chem. Mater., 2003, 15, 2815–2823 CrossRef CAS
- H. Darmstadt, C. Roy, S. Kaliaguine, T. W. Kim and R. Ryoo, Chem. Mater., 2003, 15, 3300–3307 CrossRef CAS
- W. Zhang, C. Liang, H. Sun, Z. Shen, Y. Guan, P. Ying and C. Li, Adv. Mater., 2002, 14, 1776–1778 CrossRef CAS
- A. Lu, W. Li, W. Schmidt, W. Kiefer and F. Schueth, Carbon, 2004, 42, 2939–2948 CrossRef CAS
- K. P. Gierszal and M. Jaroniec, Chem. Commun., 2004, 2576–2577 RSC
- H. I. Lee, C. Pak, C. H. Shin, H. Chang, D. Seung, J. E. Yie and J. M. Kim, Chem. Commun., 2005, 6035–6037 RSC
- T. W. Kim, F. Kleitz, B. Paul and R. Ryoo, J. Am. Chem. Soc., 2005, 127, 7601–7610 CrossRef CAS
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC
- X. Y. Liu, B. Z. Tian, C. Z. Yu, F. Gao, S. H. Xie, B. Tu, R. C. Che, L. M. Peng and D. Y. Zhao, Angew. Chem., Int. Ed., 2002, 41, 3876–3878 CrossRef CAS
- S. Che, A. E. Garcia-Bennett, X. Liu, R. P. Hodgkins, P. A. Wright, D. Zhao, O. Terasaki and T. Tatsumi, Angew. Chem., Int. Ed., 2003, 42, 3930–3934 CrossRef CAS
- H. Yang, Q. Shi, X. Liu, S. Xie, D. Jiang, F. Zhang, C. Yu, B. Tu and D. Zhao, Chem. Commun., 2002, 2842–2843 RSC
- B. Z. Tian, S. N. Che, Z. Liu, X. Y. Liu, W. B. Fan, T. Tatsumi, O. Terasaki and D. Y. Zhao, Chem. Commun., 2003, 2726–2727 RSC
- M. Sevilla, S. Alvarez and A. B. Fuertes, Microporous Mesoporous Mater., 2004, 74, 49–58 CrossRef CAS
- S. S. Kim, T. R. Pauly and T. J. Pinnavaia, Chem. Commun., 2000, 1661–1662 RSC
- H. C. Li, Y. Sakamoto, Y. S. Li, O. Terasaki, M. Thommes and S. A. Che, Microporous Mesoporous Mater., 2006, 95, 193–199 CrossRef CAS
- Y. Xia and R. Mokaya, Chem. Mater., 2005, 17, 1553–1560 CrossRef CAS
- T. Kim, R. Ryoo, K. P. Gierszal, M. Jaroniec, L. A. Solovyov, Y. Sakamoto and O. Terasaki, J. Mater. Chem., 2005, 15, 1560–1571 RSC
- W. Guo, F. Su and X. S. Zhao, Carbon, 2005, 43, 2423–2426 CrossRef CAS
- S. J. Han, K. Sohn and T. Hyeon, Chem. Mater., 2000, 12, 3337–3341 CrossRef CAS
- S. Han and T. Hyeon, Chem. Commun., 1999, 1955–1956 RSC
- S. Han, K. T. Lee, S. M. Oh and T. Hyeon, Carbon, 2003, 41, 1049–1056 CrossRef CAS
- S. J. Han, S. Kim, H. Lim, W. Y. Choi, H. Park, J. Yoon and T. Hyeon, Microporous Mesoporous Mater., 2003, 58, 131–135 CrossRef CAS
- Z. Li and M. Jaroniec, Carbon, 2001, 39, 2080–2082 CrossRef CAS
- Z. J. Li and M. Jaroniec, Chem. Mater., 2003, 15, 1327–1333 CrossRef CAS
- K. P. Gierszal and M. Jaroniec, J. Am. Chem. Soc., 2006, 128, 10026–10027 CrossRef CAS
- J. Jang and B. Lim, Adv. Mater., 2002, 14, 1390–1393 CrossRef CAS
- J. Jang, B. Lim and M. Choi, Chem. Commun., 2005, 4214–4216 RSC
- J. E. Hampsey, Q. Y. Hu, L. Rice, J. B. Pang, Z. W. Wu and Y. F. Lu, Chem. Commun., 2005, 3606–3608 RSC
- J. Lee, J. Kim and T. Hyeon, Chem. Commun., 2003, 1138–1139 RSC
- J. Lee, K. Sohn and T. Hyeon, J. Am. Chem. Soc., 2001, 123, 5146–5147 CrossRef CAS
- J. Lee, K. Sohn and T. Hyeon, Chem. Commun., 2002, 2674–2675 RSC
- Y. Oda, K. Fukuyama, K. Nishikawa, S. Namba, H. Yoshitake and T. Tatsumi, Chem. Mater., 2004, 16, 3860–3866 CrossRef CAS
- S. B. Yoon, J. Y. Kim and J. Yu, Chem. Commun., 2002, 1536–1537 RSC
- J. Kim, J. Lee and T. Hyeon, Carbon, 2004, 42, 2711–2719 CrossRef CAS
- J. Lee, J. Kim, Y. Lee, S. Yoon, S. M. Oh and T. Hyeon, Chem. Mater., 2004, 16, 3323–3330 CrossRef CAS
- S. S. Kim, D. K. Lee, J. Shah and T. J. Pinnavaia, Chem. Commun., 2003, 1436–1437 RSC
- D. Kawashima, T. Aihara, Y. Kobayashi, T. Kyotani and A. Tomita, Chem. Mater., 2000, 12, 3397–3401 CrossRef CAS
- I. Moriguchi, Y. Koga, R. Matsukura, Y. Teraoka and M. Kodama, Chem. Commun., 2002, 1844–1845 RSC
- J. Jang, B. Lim and M. Choi, Chem. Commun., 2005, 4214–4216 RSC
- B. Han, W. Zhou and A. Sayari, J. Am. Chem. Soc., 2003, 125, 3444–3445 CrossRef CAS
- J. Eric Hampsey, Q. Hu, L. Rice, J. Pang, Z. Wu and Y. Lu, Chem. Commun., 2005, 3606–3608 RSC
- S. Tanaka, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Commun., 2005, 2125–2127 RSC
- J. Pang, V. T. John, D. A. Loy, Z. Yang and Y. Lu, Adv. Mater., 2005, 17, 704–707 CrossRef CAS
- Z. X. Yang, Y. D. Xia and R. Mokaya, J. Mater. Chem., 2006, 16, 3417–3425 RSC
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS
- C. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef CAS
- F. Zhang, Y. Meng, D. Gu, Y. Yan, C. Yu, B. Tu and D. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CrossRef CAS
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS
- Y. Huang, H. Q. Cai, T. Yu, F. Q. Zhang, F. Zhang, Y. Meng, D. Gu, Y. Wan, X. L. Sun, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2007, 46, 1089–1093 CrossRef CAS
- Y. H. Deng, T. Yu, Y. Wan, Y. F. Shi, Y. Meng, D. Gu, L. J. Zhang, Y. Huang, C. Liu, X. J. Wu and D. Y. Zhao, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS
- T. Kim, I. Park and R. Ryoo, Angew. Chem., Int. Ed., 2003, 42, 4375–4379 CrossRef CAS
- C. H. Kim, D. K. Lee and T. J. Pinnavaia, Langmuir, 2004, 20, 5157–5159 CrossRef CAS
- A. B. Fuertes and S. Alvarez, Carbon, 2004, 42, 3049–3055 CrossRef CAS
- A. B. Fuertes and T. A. Centeno, J. Mater. Chem., 2005, 15, 1079–1083 RSC
- H. Yang, Y. Yan, Y. Liu, F. Zhang, R. Zhang, Y. Meng, M. Li, S. Xie, B. Tu and D. Zhao, J. Phys. Chem. B, 2004, 108, 17320–17328 CrossRef CAS
- Y. Xia, Z. Yang and R. Mokaya, Stud. Surf. Sci. Catal., 2005, 156, 565–572 Search PubMed
- Y. Xia, Z. Yang and R. Mokaya, Chem. Mater., 2006, 18, 140–148 CrossRef CAS
- F. Su, J. Zeng, X. Bao, Y. Yu, J. Y. Lee and X. S. Zhao, Chem. Mater., 2005, 17, 3960–3967 CrossRef CAS
- J. S. Lee, S. H. Joo and R. Ryoo, J. Am. Chem. Soc., 2002, 124, 1156–1157 CrossRef CAS
- H. I. Lee, J. H. Kim, D. J. You, J. E. Lee, J. M. Kim, W. S. Ahn, C. Pak, S. H. Joo, H. Chang and D. Seung, Adv. Mater., 2008, 20, 757–762 CrossRef CAS
- S. Tanaka, Y. Katayama, M. P. Tate, H. W. Hillhouse and Y. Miyake, J. Mater. Chem., 2007, 17, 3639–3645 RSC
- Y. Yan, F. Q. Zhang, Y. Meng, B. Tu and D. Y. Zhao, Chem. Commun., 2007, 2867–2869 RSC
- F. Zhang, Y. Meng, D. Gu, Y. Yan, Z. Chen, B. Tu and D. Zhao, Chem. Mater., 2006, 18, 5279–5288 CrossRef CAS
- F. Q. Zhang, D. Gu, T. Yu, F. Zhang, S. H. Xie, L. J. Zhang, Y. H. Deng, Y. Wan, B. Tu and D. Y. Zhao, J. Am. Chem. Soc., 2007, 129, 7746–7747 CrossRef CAS
- C. Vix-Guterl, S. Boulard, J. Parmentier, J. Werckmann and J. Patarin, Chem. Lett., 2002, 1062–1063 CrossRef CAS
- A. B. Fuertes and D. M. Nevskaia, J. Mater. Chem., 2003, 13, 1843–1846 RSC
- C. Yu, J. Fan, B. Tian, D. Zhao and G. D. Stucky, Adv. Mater., 2002, 14, 1742–1745 CrossRef CAS
- A. Kasuya, H. Takahashi, Y. Saito, T. Mitsugashira, T. Shibayama, Y. Shiokawa, I. Satoh, M. Fukushima and Y. Nishina, Mater. Sci. Eng., A, 1996, 217–218, 50–53 CrossRef
- Y. S. Li, Y. Q. Yang, J. L. Shi and M. L. Ruan, Microporous Mesoporous Mater., 2008, 112, 597–602 CrossRef CAS
- S. B. Yoon, K. Sohn, J. Y. Kim, C. H. Shin, J. S. Yu and T. Hyeon, Adv. Mater., 2002, 14, 19–21 CrossRef CAS
- J. S. Yu, S. B. Yoon, Y. J. Lee and K. B. Yoon, J. Phys. Chem. B, 2005, 109, 7040–7045 CrossRef CAS
- J. Pang, X. Li, D. Wang, Z. Wu, V. T. John, Z. Yang and Y. Lu, Adv. Mater., 2004, 16, 884–886 CrossRef CAS
- B. C. Chen, H. P. Lin, M. C. Chao, C. Y. Mou and C. Y. Tang, Adv. Mater., 2004, 16, 1657–1661 CrossRef CAS
- M. L. Lin, C. C. Huang, M. Y. Lo and C. Y. Mou, J. Phys. Chem. C, 2008, 112, 867–873 CrossRef CAS
- A. Taguchi, J. H. Smatt and M. Linden, Adv. Mater., 2003, 15, 1209–1211 CrossRef CAS
- A. H. Lu, J. H. Smatt, S. Backlund and M. Linden, Microporous Mesoporous Mater., 2004, 72, 59–65 CrossRef CAS
- A. H. Lu, J. H. Smatt and M. Linden, Adv. Funct. Mater., 2005, 15, 865–871 CrossRef CAS
- Z. Shi, Y. Feng, L. Xu, S. Da and Y. Liu, Mater. Chem. Phys., 2006, 97, 472–475 CrossRef CAS
- Z. Shi, Y. Feng, L. Xu, S. Da and M. Zhang, Carbon, 2003, 41, 2677–2679 CrossRef CAS
- S. Alvarez, J. Esquena, C. Solans and A. B. Fuertes, Adv. Eng. Mater., 2004, 6, 897–899 CrossRef CAS
- X. Q. Wang, K. N. Bozhilov and P. Y. Feng, Chem. Mater., 2006, 18, 6373–6381 CrossRef CAS
- Y. Xia and R. Mokaya, J. Phys. Chem. C, 2007, 111, 10035–10039 CrossRef CAS
- Y. S. Hu, P. Adelhelm, B. M. Smarsly, S. Hore, M. Antonietti and J. Maier, Adv. Funct. Mater., 2007, 17, 1873–1878 CrossRef CAS
- Z. Y. Wang, F. Li and A. Stein, Nano Lett., 2007, 7, 3223–3226 CrossRef CAS
- G. W. Zhao, J. P. He, C. X. Zhang, J. H. Zhou, X. Chen and T. Wang, J. Phys. Chem. C, 2008, 112, 1028–1033 CrossRef CAS
- K. Wang, W. Zhang, R. Phelan, M. A. Morris and J. D. Holmes, J. Am. Chem. Soc., 2007, 129, 13388–13389 CrossRef CAS
- D. J. Cott, N. Petkov, M. A. Morris, B. Platschek, T. Bein and J. D. Holmes, J. Am. Chem. Soc., 2006, 128, 3920–3921 CrossRef CAS
- J. T. Chen, K. Shin, J. M. Leiston-Belanger, M. F. Zhang and T. P. Russell, Adv. Funct. Mater., 2006, 16, 1476–1480 CrossRef CAS
- J. Fan, S. W. Boettcher, C. K. Tsung, Q. Shi, M. Schierhorn and G. D. Stucky, Chem. Mater., 2008, 20, 909–921 CrossRef CAS
- Y. Y. Wu, T. Livneh, Y. X. Zhang, G. S. Cheng, J. F. Wang, J. Tang, M. Moskovits and G. D. Stucky, Nano Lett., 2004, 4, 2337–2342 CrossRef CAS
- Y. Y. Wu, G. S. Cheng, K. Katsov, S. W. Sides, J. F. Wang, J. Tang, G. H. Fredrickson, M. Moskovits and G. D. Stucky, Nat. Mater., 2004, 3, 816–822 CrossRef CAS
- G. S. Chai, S. B. Yoon, J. S. Yu, J. H. Choi and Y. E. Sung, J. Phys. Chem. B, 2004, 108, 7074–7079 CrossRef CAS
- J. S. Yu, S. B. Yoon and G. S. Chai, Carbon, 2001, 39, 1442–1446 CrossRef CAS
- Z. B. Lei, Y. G. Zhang, H. Wang, Y. X. Ke, J. M. Li, F. Q. Li and J. Y. Xing, J. Mater. Chem., 2001, 11, 1975–1977 RSC
- F. B. Su, X. S. Zhao, Y. Wang, J. H. Zeng, Z. C. Zhou and J. Y. Lee, J. Phys. Chem. B, 2005, 109, 20200–20206 CrossRef CAS
- G. S. Chai, I. S. Shin and J. S. Yu, Adv. Mater., 2004, 16, 2057–2061 CrossRef CAS
- S. Kang, J. S. Yu, M. Kruk and M. Jaroniec, Chem. Commun., 2002, 1670–1671 RSC
- M. W. Perpall, K. P. U. Perera, J. DiMaio, J. Ballato, S. H. Foulger and D. W. Smith, Langmuir, 2003, 19, 7153–7156 CrossRef CAS
- P. Adelhelm, Y. S. Hu, L. Chuenchom, M. Antonietti, B. M. Smarsly and J. Maier, Adv. Mater., 2007, 19, 4012–4017 CrossRef CAS
- C. D. Liang and S. Dai, Chem. Mater., 2009, 21, 2115–2124 CrossRef CAS
- S. W. Woo, K. Dokko, K. Sasajima, T. Takei and K. Kanamura, Chem. Commun., 2006, 4099–4101 RSC
- Y. H. Deng, C. Liu, T. Yu, F. Liu, F. Q. Zhang, Y. Wan, L. J. Zhang, C. C. Wang, B. Tu, P. A. Webley, H. T. Wang and D. Y. Zhao, Chem. Mater., 2007, 19, 3271–3277 CrossRef CAS
- Z. Y. Wang, E. R. Kiesel and A. Stein, J. Mater. Chem., 2008, 18, 2194–2200 RSC
- Z. Y. Wang and A. Stein, Chem. Mater., 2008, 20, 1029–1040 CrossRef CAS
- S. B. Yoon, G. S. Chai, S. K. Kang, J. S. Yu, K. P. Gierszal and M. Jaroniec, J. Am. Chem. Soc., 2005, 127, 4188–4189 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2010 |