Magnetic Cu–Ni (core–shell) nanoparticles in a one-pot reaction under microwave irradiation†
Tomohisa
Yamauchi
a,
Yasunori
Tsukahara
*a,
Takao
Sakata
b,
Hirotaro
Mori
b,
Takeshi
Yanagida
c,
Tomoji
Kawai
c and
Yuji
Wada
*ad
aGraduate School of Engineering, Osaka University, 2-1 Yamada-oka Suita, Osaka 565-0871, Japan. E-mail: ytsuka@jrl.eng.osaka-u.ac.jp; Fax: +81-6-6878-2538; Tel: +81-6-6878-2538
bResearch Center for Ultra-High Voltage Electron Microscopy, Osaka University, 7-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan
cDivision of Advanced Materials Science and Technology, The Institute of Scientific and Industrial Research, Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan
dDepartment of Applied Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-12 Ookayama, Meguro, Tokyo 152-8552, Japan. E-mail: yuji-w@apc.titech.ac.jp
First published on 9th January 2010
Abstract
We successfully prepared face-centered cubic (fcc) Cu–Ni (core–shell) nanoparticles by intramolecular reduction of formate complexes of Cu2+ and Ni2+ with long-chain amine ligands in a one-pot reaction within an extremely short time realized only under microwave irradiation. Observation by an HAADF-STEM technique showed that the nanostructure in one particle consisted of a Ni-rich shell and a Cu-rich core. Cu4Ni6 nanoparticles with an average size of 11.7 nm were comprised of a Cu core with a diameter of ca. 6.0 nm, a Ni shell ca. 1.6 nm thick and a 0.9 nm thick interlayer of mixed Cu–Ni alloy between the Cu core and the Ni shell. Both the oxidation characteristics and the magnetic properties were dramatically affected by the molar ratios of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni in the Cu–Ni nanoparticles. The magnetization of Cu3Ni7 and Cu4Ni6 comprised of a diamagnetic Cu-rich core, ferromagnetic Ni-rich shell and antiferromagnetic NiO-rich layer on the particle surface showed an exchange bias (209 and 143 Oe, respectively).
:Ni in the Cu–Ni nanoparticles. The magnetization of Cu3Ni7 and Cu4Ni6 comprised of a diamagnetic Cu-rich core, ferromagnetic Ni-rich shell and antiferromagnetic NiO-rich layer on the particle surface showed an exchange bias (209 and 143 Oe, respectively).
Introduction
Nanoparticles of ferromagnetic metals, such as Fe, Co, and Ni, are used in catalysts, permanent magnets, magnetic fluids and magnetic recording media.1 The physical and chemical properties of nanoparticles depend on their size and shape.2 Currently, novel bimetallic nanoparticles with chemical and physical properties superior to those of monometallic nanoparticles have attracted theoretical and practical interest.3 Among these novel bimetallic materials, Cu–Ni nanoparticles have been widely used as catalysts,4 hyperthermic magnetic fluids,5 electrode materials in solid oxide fuel cells, in multilayer ceramic capacitors (MLCC),6 and as condenser tubing in marine applications.7 Cu–Ni alloy nanoparticles are used in place of monometallic Ni or Cu due to their chemical stability and mechanical properties.Generally speaking, three main types of mixing patterns for Cu–Ni alloy nanoparticles can be identified such as mixed Cu–Ni alloy, subcluster segregated alloy and core–shell segregated alloy.8 In the case of the mixed Cu–Ni alloy, Cu and Ni atoms are randomly dispersed without any order through the entire particle as a solid solution. The core–shell segregated alloy consists of a shell of either Cu or Ni atoms surrounding a core of the other. In this paper, the mixed Cu–Ni alloy is described as the Cu–Ni alloy for short.
The Cu–Ni system is especially noted for complete liquid and solid solubility,9 because of the small lattice size mismatch between Cu and Ni (3.62 Å and 3.54 Å, respectively) and the small positive enthalpies of solution for Cu in Ni and Ni in Cu.10 Cu–Ni alloy nanoparticles have been synthesized in both physical and chemical processing methods, as described below. For further improvements of physical and chemical properties, the preparation of Cu–Ni nanoparticles with core–shell structures still remains a significant challenge.
In physical processing methods alloy nanoparticles have been synthesized using mechanical alloying11 and by laser ablating targets prepared by cold-pressing powder mixtures of Cu and Ni under high temperatures (above 1273 K) and long processing times.12 At high temperatures, Cu–Ni alloy nanoparticles with Cu surface enrichment were obtained because Cu atoms have not only a large rate of diffusion in Ni13 but also a surface energy (σ(111) = 69.5 kJ mol−1) that is lower than that of Ni (80 kJ mol−1).14 These Cu–Ni alloy nanoparticles with Cu surface enrichment are easily oxidized in air. In order to suppress the oxidation of Cu components, a new method for the preparation of Cu–Ni nanoparticles with a Ni-rich shell is highly desirable.
Even in the chemical processing methods at lower temperatures—as with the physical processing methods—only the alloy nanoparticles were obtained. Each reduction (Cu2+ or Ni2+) has a unique redox potential (Ni2+ + 2e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) Ni, −0.257 V; Cu2+ + 2e− Cu, 0.342 V) and a unique reduction temperature. Cu–Ni nanoparticles are more likely to be produced through the following heterogeneous nucleation process due to the difference in those reduction temperatures. At first, Cu nanoparticles are produced at lower temperatures and then heterogeneous nucleation of Ni particles on the surface boundaries of Cu nanoparticles occurs much more often than homogeneous nucleation of the independent Ni particles.15 Therefore, Cu–Ni nanoparticles with Ni surface enrichment are more likely to be produced with the liquid-phase methods at lower temperatures. However, in previous reports using several liquid-phase methods, such as electrochemical deposition,16 microemulsion synthesis,17 sol–gel methods,18 various chemical reduction methods,19 polyol methods,15a,20 and hydrothermal reactions,6c,21 the relatively homogeneous Cu–Ni alloy was obtained even at lower temperatures because of the long reaction times. Ferrando et al. speculated that this mixed alloying could be attributed to low-temperature surface melting and appreciable Ni/Cu diffusion.8 The authors concluded that this speculation as to the cause of alloying was correct. Therefore, an extremely short reaction time was required for the suppression of the surface diffusion of Cu atoms and the preparation of Cu–Ni nanoparticles with Ni surface enrichment.
Ni, −0.257 V; Cu2+ + 2e− Cu, 0.342 V) and a unique reduction temperature. Cu–Ni nanoparticles are more likely to be produced through the following heterogeneous nucleation process due to the difference in those reduction temperatures. At first, Cu nanoparticles are produced at lower temperatures and then heterogeneous nucleation of Ni particles on the surface boundaries of Cu nanoparticles occurs much more often than homogeneous nucleation of the independent Ni particles.15 Therefore, Cu–Ni nanoparticles with Ni surface enrichment are more likely to be produced with the liquid-phase methods at lower temperatures. However, in previous reports using several liquid-phase methods, such as electrochemical deposition,16 microemulsion synthesis,17 sol–gel methods,18 various chemical reduction methods,19 polyol methods,15a,20 and hydrothermal reactions,6c,21 the relatively homogeneous Cu–Ni alloy was obtained even at lower temperatures because of the long reaction times. Ferrando et al. speculated that this mixed alloying could be attributed to low-temperature surface melting and appreciable Ni/Cu diffusion.8 The authors concluded that this speculation as to the cause of alloying was correct. Therefore, an extremely short reaction time was required for the suppression of the surface diffusion of Cu atoms and the preparation of Cu–Ni nanoparticles with Ni surface enrichment.
Here we propose that the suppression of the surface diffusion of Cu atoms could be achieved by shortening the reaction time to obtain Cu–Ni (core–shell) nanoparticles. A microwave-assisted method for the present study was chosen for the following reasons. Reactants are directly and quickly heated under microwave irradiation through the interaction of the oscillating electric and magnetic fields with the substances and then a reaction solution is heated uniformly in a vessel. Therefore, nucleus growth throughout the entire reaction vessel is simultaneous and homogeneous, and particles with a narrow size distribution can be obtained within a short time.22 Recently, monodispersed Ag, Cu and Ag core–Cu shell nanoparticles with a narrow size distribution were prepared within a short time using a microwave-assisted method.23 Rapid heating and a short reaction time under microwave irradiation would be effective for the suppression of the surface diffusion of Cu atoms to obtain the monodispersed Cu–Ni (core–shell) nanoparticles.
Monodispersed Ni nanoparticles were rapidly prepared via the intramolecular reduction of Ni2+ in a formate complex with long-chain amine ligands at 463 K under microwave irradiation.24 Ligation of the long-chain amine lowered the reaction temperatures by decreasing the energy barrier required for nickel formate complexes rather than for their neat formate salts. The size of the obtained particles was controlled through the use of various long-chain amine ligands. During the intramolecular reduction of formate complexes, the formate ion acts as a reducing agent for a metal ion and ultimately decomposes to hydrogen and carbon dioxide according to the following reactions.
| 2 HCOO− → 2 CO2 + H2 + 2e− |
| M2+ + 2e− → M0 (M = Cu or Ni) |
This intramolecular reduction of copper and nickel formate complexes was selected to prepare Cu–Ni (core–shell) nanoparticles with an extremely short time under microwave irradiation.
Furthermore, novel Cu–Ni nanoparticles with core–shell structures have attracted interest in their catalytic activities, chemical and physical properties, such as oxidation characteristics and magnetic properties. Zhang et al. reported that monodispersed Cu–Ni nanoparticles with a Cu-rich core and Ni-rich region were prepared by a one-pot thermolysis approach in oleylamine/1-octadecene, using metal acetylacetonatos as precursors.25 The obtained Cu–Ni nanoparticles, especially with the composition of Cu0.5Ni0.5 had high catalytic activities in the hydrolysis of NaBH4 to generate H2. Konno et al. and Tracy et al. reported that Co and Ni nanoparticles with an oxide layer surface cover showed exchange anisotropy.26 This phenomenon was explained by an exchange anisotropy interaction at the interface between the ferromagnetic (FM) region of the metal particles and the layers of antiferromagnetic (AFM) metal oxide on the particle surface. The surface of Cu–Ni nanoparticles is easily oxidized in air. The physical properties of small nanoparticles, such as conductivity and magnetic properties, should be influenced by oxide layers on the particle surface. Detailed investigations of both a nanostructure and its oxidation characteristics are absolutely imperative for the evaluation of the performance of nanometre-sized Cu–Ni particles.
The combination of both microwave method and intramolecular reduction of formate complexes of Cu2+ and Ni2+ enabled preparation of monodispersed Cu–Ni (core–shell) nanoparticles. The composition of the Cu–Ni nanoparticles was readily controlled by changing the molar ratio of both ions used in the synthesis. Both the oxidation characteristics of the particle surface and the magnetic properties of Cu, Ni and Cu–Ni nanoparticles were examined. The effects of the metal-oxide layers on both the coercivity and hysteresis loop shift are discussed.
Experimental
Materials
Nickel(II) formate dihydrate and 1-octanol were purchased from Kishida Chemical Co., Ltd. Copper(II) formate tetrahydrate and 1-octylamine were purchased from Wako Pure Chemical Industries Co., Ltd. Oleylamine ((Z)-9-octadecenylamine) was purchased from Aldrich Co., Ltd. These reagents were used as supplied.Preparation of Cu and Ni precursors
The precursors, copper(II) or nickel(II) formate complexes with oleylamine ligands, were synthesized as described in our previous work.24 The Cu precursor was synthesized by stirring a mixture of copper(II) formate tetrahydrate (2.4 mmol) and oleylamine (12.0 mmol) at room temperature for 20 min. In the case of the Ni precursor, nickel(II) formate dihydrate (3.6 mmol) and oleylamine (18 mmol) were mixed and then heated at 393 K for 10 min. The reaction solution changed from a greenish suspension to a deep-green homogeneous solution. After allowing the nickel precursor solution to return to room temperature, the color of the solution changed to turquoise.Preparation of Cu–Ni nanoparticles
Microwave heating was carried out by use of a multi-mode 2.45- GHz microwave apparatus operated at 1.25 kW (Micro Denshi Co., Ltd). The temperature of the reaction solution was measured using a fiber-optic thermometer (AMOTH TM−5886, Anritsu Meter Co., Ltd.), which was inserted directly into the solution. The agitation speed was maintained at 200 rpm using an agitator motor (EYELA MAZALA-Z, Tokyo Rikakikai Co., Ltd.) and a glass rod equipped with a polytetrafluoroethylene (PTFE) rotor blade. The Cu–Ni nanoparticles with a molar ratio of [Cu]:[Ni] = 4:6, were prepared as follows. The Cu and Ni precursors, prepared as described above (Cu2+ 2.4 mmol, Ni2+ 3.6 mmol), were added together to 1-octanol (60 ml). This solution was heated in a quartz vessel at a rate of 40 K min−1 and then allowed to stand at 463 K for 10 min under bubbling nitrogen gas. The color of the solution readily changed to black at 463 K. The reaction solution was then cooled to room temperature within 5 min by using an ice bath. The resulting particles were centrifuged, washed in methanol to remove residual long-chain amines, and dried under vacuum at 334 K for 4 h. Black Cu–Ni nanoparticles were obtained (denoted as Cu4Ni6). The temperature profiles of the reaction plotted against time and microwave (MW) power are shown in Fig. S1 of the ESI.† Cu–Ni nanoparticles prepared using other molar ratios, i.e., [Cu]:[Ni] = 3:7, 5:5 and 8:2 (denoted as Cu3Ni7, Cu5Ni5 and Cu8Ni2), were obtained under the same reaction conditions.
Preparation of Cu and Ni nanoparticles
Cu or Ni precursors prepared from their formate salt (5 mmol) and oleylamine (50 mmol) were added to 1-octanol (60 ml). Each solution was heated at a rate of 40 K min−1 under microwave irradiation and allowed to stand at either 433 K [Cu] or 463 K [Ni] for 10 min under bubbling nitrogen gas. Cu and Ni nanoparticles were obtained (denoted as Cu-1 and Ni-1). Other Ni nanoparticles of a different particle size (denoted as Ni-2) were obtained under the same reaction conditions except for the concentration of Ni precursor in octanol solution (Ni formate salt; 0.5 mmol, oleylamine; 5.0 mmol and octanol; 60 ml). The TEM image of Ni-2 is shown in Fig. S2 of the ESI.† The average particle sizes of Ni-1 and Ni-2 were 43.0 nm24 and 20.6 nm, respectively. Another type of Cu nanoparticles, prepared with 1-octylamine (C8H19N) (denoted as Cu-2), was obtained under the same reaction conditions. The crystallite sizes of Cu-1 (oleylamine, C18H37N) and Cu-2 (octylamine, C8H19N) were calculated as 5.3 nm and 24.0 nm from the peaks of the (1 1 1) planes of fcc Cu by XRD measurement using a Scherrer's equation modified for spherical particles.27 While isolation of Cu-2 (octylamine, C8H19N) by centrifugation gave a brown powder of Cu nanoparticles, Cu-1 (oleylamine, C18H37N) could not be isolated as pure Cu powder. The color of one octanol solution of Cu-1 was rapidly changed from brown to black, when exposed to air atmosphere in the washing process, indicating Cu-1 was easily oxidized in air atmosphere. The TEM images and XRD patterns of Cu-1 and Cu-2 are shown in Figs. S3 and S4 of the ESI,† respectively.Instruments
The size and morphology of the Cu–Ni nanoparticles were characterized using a transmission electron microscope (TEM) operated at 200 kV with a Hitachi H-800 (Hitachi High-Technologies Co.). The solution was dropped onto a copper grid coated with a carbon film, and the grid was dried under vacuum. The compositions of the Cu3Ni7, Cu4Ni6, Cu5Ni5 and Cu8Ni2 nanoparticles were determined using an energy dispersive X-ray spectrometer (EDS) of EMAX-7000Q type attached to the TEM. Samples were placed on a holey carbon film supported on a molybdenum grid. The crystal phases of the powders were analyzed using a MultiFlex (Rigaku Co.) with a Cu Kα radiation source in the range of the 2θ Bragg angles = 20–120° at 40 kV and 40 mA. The melting temperatures of the samples and the amount of surface modifying agent on the surface of the nanoparticles were determined by thermogravimetric and differential thermal analyses (TG-DTA), respectively, using a Thermo plus EVO/TG-DTA (Rigaku Co.). TG-DTA was performed at a heating rate of 20 °C min−1 in a nitrogen atmosphere at a flow rate of 800 mL min−1. The oxidation characteristics of Cu, Ni and Cu–Ni nanoparticles were characterized using X-ray photoelectron spectroscopy (XPS) with an Mg Kα radiation source operated at 12 kV and 20 mA with a JPS-9010MC spectrometer (JASCO Co.). Magnetic susceptibility data were obtained in applied fields ranging between −30 and 30 kOe using a SQUID susceptometer (MPMS-5S, Quantum Design Co.).Results
Identification of the structure of Cu–Ni nanoparticles
The crystal phase and metallic composition of the obtained powder were determined by the powder X-ray diffraction (PXRD) and TEM-EDS analyses. The XRD pattern of the Cu4Ni6 nanoparticle sample is shown in Fig. 1. The 2θ angles of the characteristic reflections corresponding to the (111) and (200) planes of bulk fcc metals are 43.30, 50.43° (Cu) and 44.4, 51.8° (Ni) according to JCPDS file 4-0836 (Cu) and 4-0850 (Ni), respectively. The XRD reflection spectrum of Cu4Ni6 was observed as the same fcc pattern as those of bulk fcc Cu and Ni. The two peaks of the (111) and (200) planes were observed at 2θ = 43.76 and 50.84°, respectively. The 2θ angles of these planes of Cu4Ni6 were larger than those of bulk fcc Cu and smaller than those of bulk fcc Ni. The XRD reflection spectra of Cu3Ni7, Cu5Ni5 and Cu8Ni2 were observed as the same fcc pattern as that of Cu4Ni6. The reflections corresponding to the (111) planes of Cu3Ni7, Cu5Ni5 and Cu8Ni2 observed at 44.06, 43.55 and 43.51°, respectively, were shifted to larger angles as the Ni content of the Cu–Ni nanoparticles increased. The EDS spectrum of Cu4Ni6 is shown in Fig. S5 of the ESI.† Only Ni and Cu peaks were detected in the spectrum, with the exception of Mo peaks, due to the TEM Mo grid supporting the sample. The compositions determined by the EDS spectra of all types of nanoparticles are listed in Table 1. The compositions of the Cu–Ni nanoparticles were coincident with the molar ratios [Cu2+]![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) :[Ni2+] used for the synthesis.
:[Ni2+] used for the synthesis.
| Sample | Particles sizea/nm | Metal compositionb | Weight lossc/wt (%) | |
|---|---|---|---|---|
| Cu/mol (%) | Ni/mol (%) | |||
| a Lengths of major axes are listed. b Metal compositions were calculated based on the Ni Kα and Cu Kα intensities in the EDS spectra. c Surface-modifying agent contents were derived from TG measurements. | ||||
| Cu3Ni7 | 14.6 ± 1.4 | 31.5 | 68.5 | 9.8 |
| Cu4Ni6 | 11.7 ± 1.4 | 39.3 | 60.7 | 10.0 |
| Cu5Ni5 | 12.9 ± 1.5 | 55.5 | 44.5 | 17.3 |
| Cu8Ni2 | 21.4 ± 3.2 | 80.3 | 19.7 | 11.2 |
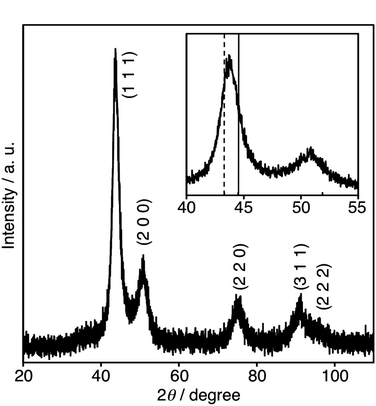 | ||
| Fig. 1 XRD pattern of Cu4Ni6. The pattern enlarged in the 40° to 55° range is shown in the inset. The characteristic reflections corresponding to the (111) planes of bulk fcc Ni (solid line) and fcc Cu (dotted line) are shown in the inset (JCPDS files 4-0850 (Ni) and 4-0836 (Cu)). | ||
TG-DTA measurements were carried out to determine both melting points and organic component contents on the surface of the Cu–Ni nanoparticles. The weight-loss curves and heat values of both Cu8Ni2 and Cu3Ni7 nanoparticles are shown in Fig. S6. The melting points measured by DTA are plotted in Fig. S7, showing a positive linear relationship with the Ni content. This relationship showed the same behavior with the melting points of the bulk well-mixed Cu–Ni alloy. The contents of the long-chain amine (oleylamine) in all samples were calculated based on the weight-loss, and are listed in Table 1. In the case of Cu4Ni6 nanoparticles, one molecule of oleylamine occupied the surface of one particle (Cu4Ni6) in the area of 17.3 nm2. (The method for estimation is described in Appendix 1 of the ESI†).
The TEM images and particle size distributions of the Cu4Ni6 nanoparticle samples are shown in Fig. 2, respectively. The obtained nanoparticles were oblate spheroid in shape with an eccentricity of 0.91. Major and minor axes were 11.7 nm (standard deviation σ = 1.4 nm) and 4.9 nm (σ = 0.5 nm), as determined from the TEM images of Fig. 2(a) and 2(b), respectively. The particle sizes of the obtained nanoparticles are listed in Table 1. The selected area electron diffraction (SAED) pattern obtained from the entire region in Fig. 2(a) is shown in the inset of Fig. 2(a). The SAED pattern showed the fcc ring pattern of (111), (200), (220), and (311) planes. While the lattice spacing estimated from the pattern [2.000 Å (111)] was close to those of fcc Cu, Ni or Cu–Ni alloys (Cu; 2.087 Å (111), Ni; 2.035 Å (111)), the value of the lattice spacings estimated by a conventional TEM at 200 keV had a margin of error to some degree. Therefore, it was difficult to clearly specify the metallic constituents from the samples of Cu, Ni and Cu–Ni alloys by SAED analysis. Another fcc ring pattern was observed as blurry. This pattern of the (111) (*) and (220) (**) planes is shown in the inset of Fig. 2(a). The lattice spacing estimated from this pattern was coincident with those of the metal oxides (NiO, CuO or Cu2O). Identification of the metal oxide was difficult using the ring pattern of this blurry ring because both fcc NiO, CuO and Cu2O had a similar lattice spacing.28 These results clearly indicate that monodispersed Cu–Ni nanoparticles containing a small amount of metal oxides were obtained.
 | ||
| Fig. 2 TEM images of Cu4Ni6 samples prepared by methanol (a) and hexane dispersion droplets (b). Size distribution of the major axes (c). The size distribution histogram was created using the diameters of 200 randomly selected particles shown in (a). Inset of (a) is an SAED pattern from the entire image. Debye rings of fcc metal were assigned as (hkl), fcc metal oxide: (111) (*) and (220) (**). | ||
The Cu–Ni nanoparticles prepared under bubbling nitrogen gas by our method could undergo oxidation, when exposed to air at room temperature in both processes of washing and preparing the samples for the measurements such as XRD, TEM and XPS, respectively. The progress of the oxidation in air at various temperatures was examined by XRD measurement. The XRD reflection spectra of the Cu4Ni6 nanoparticle sample stored at 423 and 523 K for 1 h under an air atmosphere are shown in Figs. S8(a) and (b), respectively. In both spectra, the characteristic reflections corresponding to the (111) and (220) planes of fcc metal oxides (NiO and CuO) were observed at around 2θ = 37.2° and 62.5°, respectively. The intensities of these reflections were increased with the rise in the temperature. On the other hand, the intensities of these reflections observed for the sample right after the isolation were negligible (Fig. 1). This result indicated that the Cu–Ni nanoparticles prepared by our method were not easily oxidized at room temperature. Furthermore, the XRD pattern of a Cu4Ni6 nanoparticle sample stored for one year at room temperature under an air atmosphere [Fig. S8(c)] was unchanged from that of the same sample right after the isolation. The above results led to a conclusion that the oxidation rate of the Cu–Ni nanoparticles at room temperature was extremely slow even under air. The TEM measurements with resolution in nano region should have revealed the local presence of the metal oxides in a small amount in the Cu–Ni nanoparticle samples.
The concentrations of both Cu and Ni atoms in one particle were characterized using a high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) operated at 200 kV with a Hitachi HD-2700. A HAADF-STEM image of Cu4Ni6 is shown in Fig. 3(a). The particle sizes determined from the image agree well with those from the TEM image in Fig. 2(a). Each elemental map of Cu and Ni in the area shown in Fig. 3(a) was obtained using the EDS attached to the HAADF-STEM, shown in Fig. 3(b) (Cu) and 3(c) (Ni), respectively. Cu atoms were frequently seen in the center of the particles in the image. In contrast, the distribution of Ni atoms became concentrated at the region close to the surface of the particles. The distributions between Cu and Ni atoms in a single particle are shown in Fig. 3(d). This data was obtained by measuring a single isolated particle in a different area from the measurement field shown in Fig. 3(a), which was polluted by contaminants after mapping the elements demonstrated in Fig. 3(b) and 3(c). The scan profiles shown in Fig. 3(d) were collected by point analyses along the cross-section line on a single isolated particle using EDS. For that purpose, an electron beam with 200 kV accelerating voltage was focused down to a spot with a diameter about 0.1 nm on the specimen. Cu–Ni nanoparticles (Cu4Ni6) were comprised of a Cu core with a diameter of ca. 6.0 nm surrounded by a Ni shell, ca. 2 nm thick. These results indicated that Ni shells were overgrown on Cu cores. However, the obtained nanoparticles were not simply separated as a Cu core/Ni shell region. In the region defined as the Ni shell, the layer at a depth of ca. 1.6 nm from the outermost the shell consisted chiefly of Ni atoms. The layers at depths of 1.6 nm and 2.5 nm from the outermost shell were comprised of mixed Cu–Ni alloy—in a region where the intensities of Ni atoms were larger than those of Cu atoms. Therefore, the nanostructure was identified as a Ni-rich shell and Cu-rich core, as depicted in Fig. 5(a).
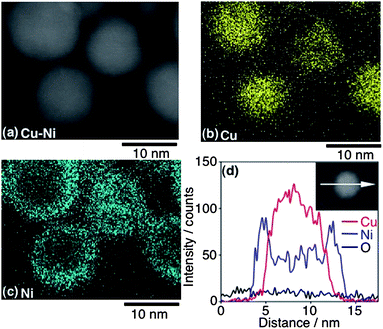 | ||
| Fig. 3 HAADF-STEM image of Cu4Ni6, Cu component (b) and Ni component (c). The scan profile along the cross-section line on a single particle is indicated in the inset (d). | ||
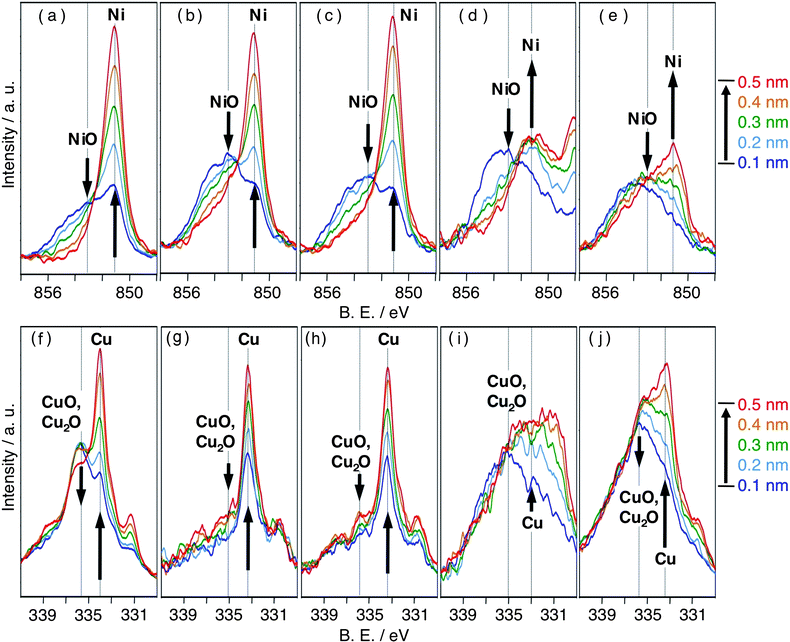 | ||
| Fig. 4 XPS spectra of Ni-1 (a), Cu-2 (f), Cu3Ni7 (b, g), Cu4Ni6 (c, h), Cu5Ni5 (d, i) and Cu8Ni2 (e, j). Figures (x, y) show Ni 2p3/2 electron spectra and Cu L3M4,5M4,5 Auger electron spectra, respectively. | ||
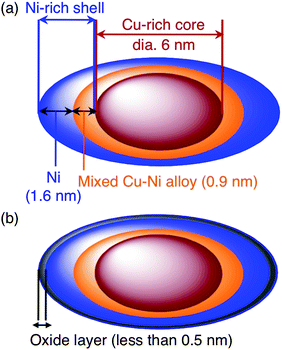 | ||
| Fig. 5 The nanostructures of Cu4Ni6 determined by HAADF-STEM (a) and XPS analyses (b), respectively. | ||
Oxidation characteristics of Cu–Ni nanoparticels (XPS)
The oxidation characteristics of the Cu–Ni nanoparticles prepared using different molar ratios of Cu:Ni were compared using XPS. The depth profiles measured between 0.1–0.5 nm from the nanoparticle surface were obtained using an ion (Ar+) sputtering technique. These surface layers at depths of between 0.1 nm and 0.5 nm were involved in the Ni-rich shell, depicted in Fig. 5(a). Therefore, the results of the depth profiles showed the oxidation state in the Ni-rich shell. Ni 2p3/2, Cu L3M4,5M4,5 Auger, and Cu 2p3/2 electron spectra are shown in Fig. 4 and Fig. S9 respectively. As shown in Fig. 4(a), the binding energies (B.E.) of Ni 2p3/2 and NiO 2p3/2 were 851 and 853 eV, respectively. In the Cu Auger spectra [Fig. 4(f)], CuO and Cu2O had the same binding energies (336 eV), which were different for Cu (334 eV). In the Cu 2p3/2 spectra (Fig. S9), Cu and Cu2O had the same binding energies (931.5 eV), which were different for CuO (932.5 eV). Therefore, each oxidation characteristic of Ni and Cu components was examined by comparing the intensities of NiO with those of Ni in the Ni 2p3/2 spectra and the intensities of mixed CuO/Cu2O with those of Cu in the Cu Auger spectra. The depth profiles of Cu3Ni7 and Cu4Ni6 showed that NiO and CuO/Cu2O were present only in negligible amounts at a depth of 0.5 nm. In contrast, the depth profiles of Cu5Ni5 and Cu8Ni2 revealed the existence of NiO and CuO/Cu2O even at a depth of 0.5 nm. Therefore, the abundance ratios of the metal oxides (NiO and CuO/Cu2O) relative to the corresponding metals increased as the Ni content of the Cu–Ni nanoparticles decreased.
Table 2 shows the relative intensities of metal oxides (NiO and CuO/Cu2O) for the corresponding metals (Ni and Cu) measured at a surface depth of 0.1 nm to 0.5 nm for each type of the nanoparticles. The relative intensities of the metal oxides were highly dependent on Ni content (atm%). When the Ni content was greater than 50 atm%, i.e., Cu3Ni7 and Cu4Ni6, the layer at a surface depth of 0.1 nm mostly consisted of Ni, Cu and NiO, while at depths greater than 0.5 nm only Ni and Cu atoms were present. On the other hand, when the Ni content was less than 50 atm%, i.e., Cu5Ni5 and Cu8Ni2, the layer at a surface depth of 0.1 nm was mostly comprised of NiO and CuO/Cu2O, and that at a depth of 0.5 nm consisted of a mixture of Ni, Cu, NiO, and CuO/Cu2O. These results indicate that the Cu3Ni7 and Cu4Ni6 nanoparticles were partially covered with a ca. 0.5 nm thick metal oxide layer, which mainly consisted of NiO. The nanostructure for Cu4Ni6 determined by both HAADF-STEM and XPS analyses are depicted in Fig. 5(b). The nanoparticles with a lower Ni content, i.e., Cu5Ni5 and Cu8Ni2, were covered with an oxide layer more than 0.5 nm thick, which consisted of mixed oxide Cu–O–Ni.
| Sample | Depth/nm | Intensities (Ni 2p3/2) | Intensities (Cu Auger) | Relative intensities | |||
|---|---|---|---|---|---|---|---|
| I Ni | I NiO | I Cu | I CuO/Cu2O | I NiO/INi | I CuO/Cu2O/ICu | ||
| Ni-1 | 0.1 | 820 | 585 | — | — | 0.71 | — |
| 0.5 | 2700 | 350 | — | — | 0.13 | — | |
| Cu3Ni7 | 0.1 | 421 | 622 | 128 | 28 | 1.48 | 0.22 |
| 0.5 | 1550 | 400 | 232 | 55 | 0.26 | 0.24 | |
| Cu4Ni6 | 0.1 | 496 | 575 | 237 | 86 | 1.16 | 0.36 |
| 0.5 | 2050 | 350 | 420 | 120 | 0.17 | 0.29 | |
| Cu5Ni5 | 0.1 | 230 | 380 | 60 | 148 | 1.65 | 2.47 |
| 0.5 | 435 | 215 | 178 | 169 | 0.49 | 0.95 | |
| Cu8Ni2 | 0.1 | 88 | 255 | 215 | 427 | 2.90 | 1.99 |
| 0.5 | 340 | 300 | 530 | 524 | 0.88 | 0.99 | |
The HRTEM image of Cu4Ni6 in Fig. 6 shows the lattice spacing on the surface of the nanoparticles. The image in Fig. 6(b) shows two lattice spacings of 0.243 and 0.207 nm, which were similar to those of the (111) and (200) planes of fcc NiO, respectively.27 The oxide layers were of low thickness and had different directions in planes. These results indicate that Cu4Ni6 nanoparticles were covered with squamiform NiO layers, as depicted in Fig. 6(c).
 | ||
| Fig. 6 HRTEM image of a Cu4Ni6 nanoparticle, original TEM (a), lattice spacings (red line; 0.243 nm, black line; 0.207 nm) (b) and graphical scheme of the particle (c). | ||
Magnetic properties
The magnetic properties of the Cu–Ni nanoparticles were measured using a SQUID susceptometer. All samples were measured as a powder. Fig. 7 shows a plot of magnetization versus applied fields for the Cu–Ni nanoparticles in zero-field cooling (ZFC) at 5 K. The saturation magnetization (σs), coercivity (Hc) and exchange bias (Heb) of Cu–Ni nanoparticles prepared using different molar ratios of Cu:Ni are listed in Table 3. The saturation magnetizations of Cu3Ni7, Cu4Ni6 and Cu5Ni5 with average particle sizes between 12–15 nm decreased as the Ni content decreased. The magnetization of Cu3Ni7 and Cu4Ni6 showed stronger ferromagnetic properties than Cu5Ni5 and Cu8Ni2. Hysteresis loops were measured at 5 K after completion of both ZFC and field cooling (FC) processes. In the FC process, the sample was cooled from 300 K to 5 K in a magnetic field, H, of 1 T. Fig. 8 shows the ZFC and FC loops obtained for Cu3Ni7. At 5 K, an asymmetric magnetic hysteresis loop and a deviation between the ZFC and FC magnetizations, which are commonly referred to as an exchange bias field (Heb), were observed.26 The Heb (= |HFC1 + HFC2|/2) was calculated to be 209 Oe, which was indicative of an exchange bias effect. Under the condition of ZFC, the Hc (= |HZFC1 − HZFC2|/2) was 638 Oe, indicating a random effect.
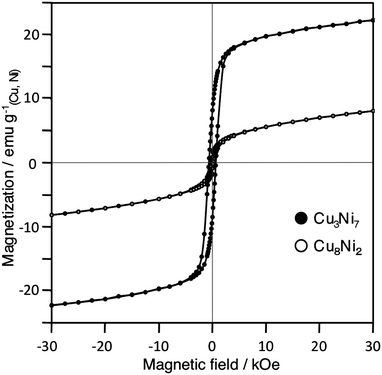 | ||
| Fig. 7 Magnetization versus applied field for Cu3Ni7 and Cu8Ni2 in ZFC at 5 K. | ||
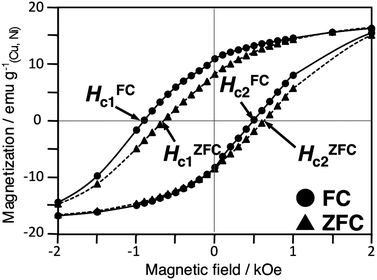 | ||
| Fig. 8 Hysteresis loops for Cu3Ni7 after both ZFC and FC at 5 K. | ||
Discussion
Mechanism of Cu–Ni (core–shell) nanoparticle production
Effect of the oxide layers of the Cu–Ni nanoparticles on the coercivities and hysteresis loop shifts
Bulk Cu–Ni alloys are known to be completely solid solutions. The density of states for Ni, Cu and Cu–Ni alloys are as follows. Ni metal (valence electrons: 10) has 0.6 electrons in the 4s band, and 5 and 4.4 electrons in the 3d↑and 3d↓ bands, respectively, with a hole of 0.6 electrons in the 3d↓ band. Therefore, Ni metal has both a spontaneous magnetization of 0.6 μB (μB:Bohr magneton) and ferromagnetic (FM) properties. Cu metal (valence electrons: 11), which has 10 electrons in the 3d band and one in the 4s band, shows diamagnetism. The Slater–Pauling curve shows that the magnetic properties of bulk Cu–Ni alloys decrease as the Cu content increases because the 0.6-electron hole in the 3d band of Ni is filled by the 4s electron of Cu.30 With the composition of the bulk Cu3.75Ni6.25 alloy, the ferromagnetic property disappears. In the case of the obtained Cu–Ni (core–shell) nanoparticles prepared in our method, the saturation magnetizations of Cu3Ni7 had weak saturation magnetization compared to that of Cu4Ni6, but remained FM (ferromagnetic) property in contrasted with the bulk Cu3.75Ni6.25 alloy. This result indicates that some of the FM Ni components remained in the nanoparticles.The effects of an oxide layer on the particle surface on hysteresis loop shift were examined. Cu and CuO have diamagnetic (DM) properties, and Ni and NiO have FM and antiferromagnetic (AFM) properties, respectively. XPS analyses of the FM Cu–Ni (core–shell) nanoparticles (Cu3Ni7 and Cu4Ni6) showed that the 0.5 nm thick outer layer of the nanoparticles consisted of a NiO-rich layer. Furthermore, HAADF-STEM analysis of Cu4Ni6 showed that these nanoparticles were comprised of a Cu-rich core with a diameter of ca. 6.0 nm, and were surrounded by a Ni-rich shell with a ca. 2.5-nm thick. These results indicated that the obtained particles had three magnetic layers:a DM Cu-rich core, a FM Ni-rich shell and an AFM NiO-rich layer on the surface. Therefore, exchange bias fields (Heb) were observed as a result of exchange coupling between three magnetic layers (DM/FM/AFM). A magnetic curve for Ni-2 with the average particle size of 20.6 nm is shown in Fig. S11. The magnetic curve for sample Ni-2 showed no Heb, which was understandable considering the relationship between the weak AFM of the very thin NiO layer and the strong FM of the Ni core compared with Cu3Ni7 and Cu4Ni6.
Conclusion
In summary, we succeeded in the rapid preparation of monodispersed Cu–Ni (core–shell) nanoparticles by intramolecular reduction of formate complexes of Cu2+ and Ni2+ with long-chain amine ligands in a one-pot reaction under microwave irradiation. The composition of the Cu–Ni nanoparticles could be easily controlled by changing the molar ratios of Cu:Ni used for the synthesis. The sizes of the resulting Cu–Ni nanoparticles were 14.6 nm (Cu3Ni7), 11.7 nm (Cu4Ni6), 12.9 nm (Cu5Ni5) and 21.9 nm (Cu8Ni2). The nanoparticles prepared using our method had different concentrations of both Cu and Ni atoms in the core and shell regions within a single particle. Observation by the HAADF-STEM technique showed the nanostructure was comprised of a Ni-rich shell and a Cu-rich core. Cu–Ni nanoparticles (Cu4Ni6) were comprised of a Cu core with a diameter of ca. 6.0 nm, a Ni shell with a ca. 1.6 nm thick and an interlayer of mixed Cu–Ni alloy 0.9 nm thick between the Cu core and the Ni shell. Both the oxidation characteristics and the magnetic properties were dramatically affected by the molar ratios of Cu:Ni in the Cu–Ni nanoparticles. The magnetization of Cu–Ni nanoparticles showed exchange bias with three different magnetic properties: DM (Cu-rich core), FM (Ni-rich shell) and AFM (NiO-rich layer on the particle surface).
Acknowledgements
This work supported financially by Nippon Steel Chemical Co., Ltd, Japan and Iwatani International Co., Ltd, Japan.References
- (a) G. Schmid, Chem. Rev., 1992, 92, 1709 CrossRef CAS; (b) K. R. Gopidas, J. K. Whitesell and M. A. Fox, Nano Lett., 2003, 3, 1757 CrossRef CAS; (c) D. J. Maxwell, J. R. Taylor and S. Nie, J. Am. Chem. Soc., 2002, 124, 9606 CrossRef CAS; (d) P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729 CrossRef CAS; (e) T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648 CrossRef CAS; (f) M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS; (g) M. M. Oliveira, E. G. Castro, C. D. Canestraro, D. Zanchet, D. Ugarte and L. S. Roman, J. Phys. Chem. B, 2006, 110, 17063 CrossRef CAS; (h) S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 30, 611; (i) W. X. Zhang, C. B. Wang and H. L. Lien, Catal. Today, 1998, 40, 387 CrossRef CAS.
- (a) D. V. Leff, P. C. Ohara, J. R. Heath and W. M. Gelbart, J. Phys. Chem., 1995, 99, 7036 CrossRef CAS; (b) C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS; (c) I. O. Sosa, C. Noguez and R. G. Barrera, J. Phys. Chem. B, 2003, 107, 6269 CrossRef CAS.
- (a) M. Chen, C.-L. Chien and P. C. Searson, Chem. Mater., 2006, 18, 1595 CrossRef CAS; (b) A. K. Salem, P. C. Searson and K. W. Leong, Nat. Mater., 2003, 2, 668 CrossRef CAS; (c) M. Heemeier, A. F. Carlsson, M. Naschitzki, M. Schmal, M. Bäumer and H.-J. Freund, Angew. Chem., Int. Ed., 2002, 41, 4073 CrossRef CAS; (d) M. Tsuji, S. Hikino, Y. Sano and M. Horigome, Chem. Lett., 2009, 38, 518 CrossRef CAS.
- (a) S. Parks, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265 CrossRef CAS; (b) Y. Li, J. Chen, L. Chang and Y. Qin, J. Catal., 1998, 178, 76e83; (c) F. M. Bautista, J. M. Campelo, A. Garcia, R. Guardeño, D. Luna and J. M. Marinas, J. Mol. Catal. A: Chem., 1996, 104, 229 CrossRef CAS; (d) T. V. Reshetenko, L. B. Avdeeva, Z. R. Ismagilov, A. L. Chuvilin and V. A. Ushakov, Appl. Catal., A, 2003, 247, 51 CrossRef CAS; (e) E. Asedegbega-Nieto, B. Bachiller-Baeza, A. Guerrero-Ruíz and I. Rodríguez-Ramos, Appl. Catal., A, 2006, 300, 120 CrossRef CAS; (f) M. Jafarian, R. B. Moghaddam, M. G. Mahjani and F. Gobal, J. Appl. Electrochem., 2006, 36, 913 CrossRef CAS.
- (a) J. Chatterjee, M. Bettge, Y. Haik and C. J. Chen, J. Magn. Magn. Mater., 2005, 293, 303 CrossRef CAS; (b) A. A. Kuznetsov, V. G. Leontiev, V. A. Brukvin, G. N. Vorozhtsov, B. Y. Kogan, O. A. Shlyakhtin, A. M. Yunin, O. I. Tsybin and O. A. Kuznetsov, J. Magn. Magn. Mater., 2007, 311, 197 CrossRef CAS.
- (a) T. Hibino, A. Hashimoto, T. Inoue, J. Tokuno, S. Yoshida and M. Sano, Science, 2000, 288, 2031 CrossRef CAS; (b) W. Songping, IEEE Trans. Compon. Packag. Technol., 2006, 29, 827 CrossRef CAS; (c) W. Songping, J. Li, N. Jing, Z. Zhenou and L. Song, Intermetallics, 2007, 15, 1316 CrossRef; (d) W. Chen, L. Li, J. Qi, Y. Wang and Z. Gui, J. Am. Ceram. Soc., 1998, 81, 2751.
- (a) W. A. Badawy, K. M. Ismail and A. M. Fathi, Electrochim. Acta, 2005, 50, 3603 CrossRef CAS; (b) R. E. Hummel and R. J. Smith, Corros. Sci., 1988, 28, 279 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845 CrossRef CAS.
- T. B. Massalki, H. Okamoto, P. R. Subramanian, Binary Alloy Phase diagrams, 2nd edn, ASM International, Metals park, OH, 1990 Search PubMed.
- C. Kittel, Introduction to Solid State Physics, 6th edn, John Wiley, New York, 1986 Search PubMed.
- S. K. Pabi, J. Joardar, I. Manna and B. S. Murty, Nanostruct. Mater., 1997, 9, 149 CrossRef CAS.
- R. P. Van Ingen, R. H. J. Fastenau and E. J. Mittenmeijer, J. Appl. Phys., 1994, 76, 1871 CrossRef CAS.
- L. A. Girifalco, Atomic Migration in Crystals, Blaisdell Publ. Co, 1964 Search PubMed.
- (a) D. S. Mainardi and P. B. Balbuena, Langmuir, 2001, 17, 2047 CrossRef CAS; (b) L. Zhu and A. E. DePristo, J. Catal., 1997, 167, 400 CrossRef CAS; (c) E. Hristova, Y. Dong, V. G. Grigoryan and M. Springborg, J. Phys. Chem. A, 2008, 112, 7905 CrossRef CAS; (d) W. F. Egelhoff, Phys. Rev. Lett., 1983, 50, 587 CrossRef CAS.
- (a) F. Bonet, S. Grugeon, L. Dupont, R. H. Urbina, C. Guéry and J. M. Tarascon, J. Solid State Chem., 2003, 172, 111 CrossRef CAS; (b) D. Kashchiev, Nucleation, Butterworth Heinemann, Oxford, 2000 Search PubMed.
- (a) H1`. Natter, M. Schmelzer and R. Hempelmann, J. Mater. Res., 1998, 13, 1186 CAS; (b) A. Foyet, A. Hauser and W. Schäfer, J. Solid State Chem., 2003, 172, 114.
- J. Feng and C.-P. Zhang, J. Colloid Interface Sci., 2006, 293, 414 CrossRef CAS.
- M. de los A. Cangiano, A. C. Carreras, M. W. Ojeda and M. del C. Ruiz, J. Alloys Compd., 2008, 458, 405 CrossRef.
- (a) C. Damle and M. Sastry, J. Mater. Chem., 2002, 12, 1860 RSC; (b) C.-H. Jung, H.-G. Lee, C.-J. Kim and S. B. Bhaduri, J. Nanopart. Res., 2003, 5, 383 CrossRef CAS.
- W.-W. Zhang, Q.-Q. Cao, J.-L. Xie, X.-M. Ren, C.-S. Lu, Y. Zhou, Y.-G. Yap and Q.-J. Meng, J. Colloid Interface Sci., 2003, 257, 237 CrossRef CAS.
- Y. D. Li, L. Q. Li, H. W. Liao and H. R. Wang, J. Mater. Chem., 1999, 9, 2675 RSC.
- (a) Y. Tsukahara, T. Nakamura, T. Kobayashi and Y. Wada, Chem. Lett., 2006, 35, 1396 CrossRef CAS; (b) J.-H. Lee, C.-K. Kim, S. Katoh and R. Murakami, J. Alloys Compd., 2001, 325, 276 CrossRef CAS; (c) F. Bondioli, A. M. Ferrari, C. Leonelli, C. Siligardi and G. C. Pellacani, J. Am. Chem. Soc., 2001, 84, 2728 CAS; (d) M. N. Nadagouda and R. S. Varma, Cryst. Growth Des., 2008, 8, 291 CrossRef CAS.
- (a) T. Yamamoto, Y. Wada, T. Sakata, H. Mori, M. Goto, S. Hibino and S. Yanagida, Chem. Lett., 2004, 33, 158 CrossRef CAS; (b) T. Nakamura, Y. Tsukahara, T. Sakata, H. Mori, Y. Kanbe, H. Bessho and Y. Wada, Bull. Chem. Soc. Jpn., 2007, 80, 224 CrossRef CAS; (c) T. Nakamura, Y. Tsukahara, T. Yamauchi, T. Sakata, H. Mori and Y. Wada, Chem. Lett., 2007, 36, 154 CrossRef CAS.
- T. Yamauchi, Y. Tsukahara, T. Sakamoto, T. Kono, M. Yasuda, A. Baba and Y. Wada, Bull. Chem. Soc. Jpn., 2009, 82, 1044 CrossRef CAS.
- Y. Zhang, W. Huang, S. E. Habas, J. N. Kuhn, M. E. Grass, Y. Yamada, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2008, 112, 12092 CrossRef CAS.
- (a) D. L. Peng, K. Sumiyama, T. Hihara, S. Yamamura and T. Konno, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3103 CrossRef CAS; (b) A. C. Johnston-Peck, J. Wang and J. B. Tracy, ACS Nano, 2009, 3, 1077 CrossRef CAS; (c) Y. D. Yao, Y. Y. Chen, M. F. Tai, D. H. Wang and H. M. Lin, Mater. Sci. Eng., A, 1996, 217–218, 281 CrossRef; (d) Y. D. Yao, Y. Y. Chen, C. M. Hsu, H. M. Lin, C. Y. Tung, M. F. Tai, D. H. Wang, K. T. Wu and C. T. Suo, Nanostruct. Mater., 1995, 6, 933 CrossRef; (e) J. Nogués and I. K. Schuller, J. Magn. Magn. Mater., 1999, 192, 203 CrossRef CAS.
- (a) A. Guinier, Theorie et Technique de la Radiocristallographie, Dunod, Paris, 1956, 482 Search PubMed; (b) H. Natter, M. Schmeltzer, M. S. Loffler, C. E. Krill, A. Fitch and R. Hempelmann, J. Phys. Chem. B, 2000, 104, 2467 CrossRef CAS; (c) R. A. Butera and D. H. Waldeck, J. Chem. Educ., 1997, 74, 115 CrossRef CAS.
- The lattice spacings of (1 1 1), (2 0 0) and (2 2 0) planes for each metal oxide were the following. {(1 1 1); 2.412 Å (NiO), 2.459 Å (CuO) and 2.427 Å (Cu2O)}, {(2 0 0); 2.089 Å (NiO), 2.130 Å (CuO) and 2.120 Å (Cu2O)}, {(2 2 0); 1.476 Å (NiO), 1.506 Å (CuO) and 1.486 Å (Cu2O)}.
- V. Rosenband and A. Gany, J. Mater. Process. Technol., 2004, 153–154, 1058 CrossRef CAS.
- S. Chikazumi, Physics of Magnetism, Wiley, New York, 1964, 73 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Time profile of both temperature and MW power during the synthesis and TEM-EDS spectrum of Cu4Ni6. TG-DTA data of Cu8Ni2 and Cu3Ni7 and melting plots for various compositions of Cu–Ni nanoparticles. Cu 2p3/2 electron spectra of various Cu–Ni nanoparticles and magnetic susceptibility of Ni-2. See DOI: 10.1039/b9nr00302a |
| This journal is © The Royal Society of Chemistry 2010 |