Liposomal squalenoyl-gemcitabine: formulation, characterization and anticancer activity evaluation
Barbara
Pili
a,
L. Harivardhan
Reddy†
a,
Claudie
Bourgaux
a,
Sinda
Lepêtre-Mouelhi
ab,
Didier
Desmaële
b and
Patrick
Couvreur
*a
aUniversité Paris-Sud XI, UMR CNRS 8612, 5 rue J.B. Clément, 92290, Châtenay-Malabry, France. E-mail: patrick.couvreur@u-psud.fr
bUniversité Paris-Sud XI, UMR CNRS 8076 Biocis, 5 rue J.B. Clément, 92290, Châtenay-Malabry, France
First published on 5th July 2010
Abstract
A new prodrug of gemcitabine, based on the covalent coupling of squalene to gemcitabine (GemSQ), has been designed to enhance the anticancer activity of gemcitabine, a nucleoside analogue active against a wide variety of tumors. In the present study, the feasibility of encapsulating GemSQ into liposomes either PEGylated or non-PEGylated has been investigated. The in vivo anticancer activity of these formulations has been tested on subcutaneous grafted L1210wt leukemia model and compared to that of free gemcitabine. The liposomal GemSQ appears to be a potential delivery system for the effective treatment of tumors.
1. Introduction
Gemcitabine is a pyrimidine antimetabolite that displays anticancer activity by inducing an S-phase arrest and inhibiting the DNA synthesis. Gemcitabine has been demonstrated to be effective in the treatment of a wide variety of solid tumors such as head and neck, colon, ovarian cancers etc., in clinical trials and has been approved for use against non-small cell lung cancer, pancreatic, bladder, and metastatic breast cancer.1–4 To be cell internalized, gemcitabine requires to be transported actively by the membrane nucleoside transporters hENT1. Intracellularly, this molecule is further phosphorylated first into monophosphate form by deoxycytidine kinase (dCk) and then into di- and active triphosphate forms by pyrimidine kinases. However, intravenously injected gemcitabine undergoes rapid metabolism through deamination into the inactive uracil derivative, hence resulting in a very short plasma half-life.5Liposomes have been employed for the encapsulation of gemcitabine with the aim to modify the drug pharmacokinetics and biodistribution and to deliver gemcitabine more efficiently to tumors. It has been found that liposomal gemcitabine exhibited greater anticancer activity than did free gemcitabine.6–9 Composition used for liposomal formulation and the preparation techniques were, however, observed to greatly influence the loading capacity of gemcitabine into liposomes and the cell penetration properties of the entrapped drug.10 But more importantly, gemcitabine which is a low-molecular weight hydrophilic molecule as well as Ara-C or 5-fluorouridine, the drugs closely related to gemcitabine, may rapidly leak out from liposomes. In order to limit this problem, various alkyl derivatives of Ara-C have been synthesized. These lipophilic pro-drugs have been encapsulated into liposomes with high efficiency and these liposomal formulations displayed in vivo antitumor activity superior to that of the pure drug.11 Similarly, a series of acyl derivatives of gemcitabine have been synthesized and encapsulated into liposomal formulations leading to modified pharmacokinetics.12 For instance, liposomes loaded with a 4-(N)-stearoyl derivative of gemcitabine showed an improved in vivo anticancer activity against HT-29 colon adenocarcinoma and KB396p nasopharyngeal carcinoma.13 Thus, liposomal prodrugs may be beneficial in terms of drug incorporation efficiency, stability, and biopharmaceutical properties.
In this context, we have synthesized a new derivative of gemcitabine by coupling the acyclic isoprenoid chain of squalene on the 4-amino function of gemcitabine. The resulting bioconjugate, i.e. 4-(N)-1,1′,2-trisnor-squalenoylgemcitabine (GemSQ), displayed more potent cytotoxicity in vitro, and exhibited considerably higher anticancer activity in vivo compared to gemcitabine against P388 and L1210wt leukemia following intravenous dosing.14–17 However, nanoassemblies of this prodrug showed considerable accumulation in organs of the reticuloendothelial system such as the spleen and liver after intravenous administration, probably due to important capture by macrophages.18 This is the reason why we performed the PEGylation of GemSQ nanoassemblies by the addition of polyethyleneglycol coupled to squalene (PEGSQ). This nanoconstruction was, however, found to be very unstable in biological media, due to the formation of unstable micelles, resulting from the solubilization of GemSQ nanoassemblies by PEGSQ in the presence of proteins.
Thus, in the present study, we have investigated the feasibility of encapsulating GemSQ into PEGylated liposomes to confer long circulating properties to this anticancer prodrug. The in vivo anticancer activity of this formulation has been tested on a subcutaneous grafted L1210wt leukemia model and compared with the activity of GemSQ encapsulated in non-PEGylated liposomes of the same composition or with free gemcitabine.
2. Experimental
2.1 Materials
1,2-Dipalmitoyl-sn-3-phosphatidylcholine (DPPC) (molecular weight 733.56, purity 99%) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG) were purchased from Avanti Polar Lipids (Alabaster, Alabama, USA) and used without further purification. Isotonic saline solution (NaCl 0.9%) was purchased from Aguettant (Lyon, France).Gemcitabine hydrochloride (2′-deoxy-2′,2′-difluorocytidine monohydrochloride; C9H11F2N3O4 HCl, β-isomer) was purchased from Sequoia Research Products (Pangbourne, UK). Squalene was purchased from Sigma-Aldrich (Saint-Quentin, Fallavier, France).
2.2 Synthesis of 4-(N)-trisnorsqualenoyl gemcitabine (GemSQ)
Gemcitabine squalene (GemSQ, Fig.1) was synthesized by covalent coupling of 1,1′,2-trisnorsqualenic acid to the 4-amino group of gemcitabine as previously described.15Briefly, to a stirred solution of 1,1′,2-trisnorsqualenoic acid (0.5 g, 1.2 mM) in anhydrous THF (3 mL) triethylamine (0.150 g, 1.5 mM) was added dropwise. The mixture was cooled to −15 °C, and a solution of ethyl chloroformate (0.135 g, 1.2 mM) in anhydrous THF (3 mL) was added dropwise. The mixture was stirred at 0 °C for 15 min, and a solution of gemcitabine hydrochloride (dFdC HCl) (0.37 g, 1.2 mmol) with triethylamine (0.24 g, 2.4 mM) in anhydrous DMF (5 mL) was added dropwise to the reaction at the same temperature. The reaction was stirred for 72 h at room temperature, and the reaction mixture was then concentrated in vacuo. Aqueous sodium hydrogen carbonate was added, and the mixture was extracted with ethyl acetate (3 × 50 mL). The combined extracts were washed with water, dried on MgSO4, and evaporated. The crude product was purified by chromatography on silica gel and eluted with CH2Cl2/MeOH/Et3N 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 then with CH2Cl2/MeOH/Et3N 100:5:1 to give pure 4-N-squalenoyl-gemcitabine (0.46 g, 57%) as an amorphous white solid: [α]D 3.1 (c 0.95, CH2Cl2); IR (neat, cm−1) ν 3500–3150, 2950, 2921, 2856, 1709, 1656, 1635, 1557, 1490, 1435, 1384, 1319, 1275, 1197, 1130, 1071, 814; 1H-NMR (300 MHz, CDCl3) δ 9.15 (s large, 1H, NHCO), 8.16 (d, 1H, J = 7.5 Hz, H6), 7.47 (d, 1H, J = 7.5 Hz, H5), 6.18 (t, 1H, J = 7.0 Hz, H1′), 5.22–5.15 (m, 5H, HVinyl), 4.49 (m, 1H, H3′), 4.86–4.09 (m, 3H, H4′, 2H5′), 2.55 (m, 2H, COCH2CH2), 2.38–2.28 (m, 2H, COCH2CH2), 2.13–1.91 (m, 16H, CH2), 1.69–1.55 (m, 18H, CDC(CH3)); 13C-NMR (100 MHz, CDCl3) δ 173.7 (CONH), 163.0 (CO), 155.8 (C), 145.4 (CH), 135.1 (C), 134.9 (2 C), 132.7 (C), 131.1 (C), 125.7 (CH), 124.4 (CH), 124.3 (CH), 124.2 (2 CH), 122.3 (t, JCF = 260 Hz, CF2), 97.7 (CH), 85.8 (m, CH), 81.6 (CH), 69.2 (t, J = 21 Hz, CH), 59.7 (CH2), 39.7 (2 CH2), 39.5 (CH2), 36.5 (CH2), 34.3 (CH2), 29.6 (CH2), 28.3 (CH2), 26.8 (CH2), 26.7 (CH2), 26.6 (CH2), 25.6 (CH3), 17.6 (CH3), 16.0 (2 CH3), 15.9 (CH3), 15.8 (CH3); MS (−ESI) m/z = 644 ([M − H]−, 100%). Anal. calcd for C36H53F2N3O5: C, 66.95, H, 8.27, N, 6.51. Found: C, 66.76, H, 8.40, N, 6.39.
:2:1 then with CH2Cl2/MeOH/Et3N 100:5:1 to give pure 4-N-squalenoyl-gemcitabine (0.46 g, 57%) as an amorphous white solid: [α]D 3.1 (c 0.95, CH2Cl2); IR (neat, cm−1) ν 3500–3150, 2950, 2921, 2856, 1709, 1656, 1635, 1557, 1490, 1435, 1384, 1319, 1275, 1197, 1130, 1071, 814; 1H-NMR (300 MHz, CDCl3) δ 9.15 (s large, 1H, NHCO), 8.16 (d, 1H, J = 7.5 Hz, H6), 7.47 (d, 1H, J = 7.5 Hz, H5), 6.18 (t, 1H, J = 7.0 Hz, H1′), 5.22–5.15 (m, 5H, HVinyl), 4.49 (m, 1H, H3′), 4.86–4.09 (m, 3H, H4′, 2H5′), 2.55 (m, 2H, COCH2CH2), 2.38–2.28 (m, 2H, COCH2CH2), 2.13–1.91 (m, 16H, CH2), 1.69–1.55 (m, 18H, CDC(CH3)); 13C-NMR (100 MHz, CDCl3) δ 173.7 (CONH), 163.0 (CO), 155.8 (C), 145.4 (CH), 135.1 (C), 134.9 (2 C), 132.7 (C), 131.1 (C), 125.7 (CH), 124.4 (CH), 124.3 (CH), 124.2 (2 CH), 122.3 (t, JCF = 260 Hz, CF2), 97.7 (CH), 85.8 (m, CH), 81.6 (CH), 69.2 (t, J = 21 Hz, CH), 59.7 (CH2), 39.7 (2 CH2), 39.5 (CH2), 36.5 (CH2), 34.3 (CH2), 29.6 (CH2), 28.3 (CH2), 26.8 (CH2), 26.7 (CH2), 26.6 (CH2), 25.6 (CH3), 17.6 (CH3), 16.0 (2 CH3), 15.9 (CH3), 15.8 (CH3); MS (−ESI) m/z = 644 ([M − H]−, 100%). Anal. calcd for C36H53F2N3O5: C, 66.95, H, 8.27, N, 6.51. Found: C, 66.76, H, 8.40, N, 6.39.
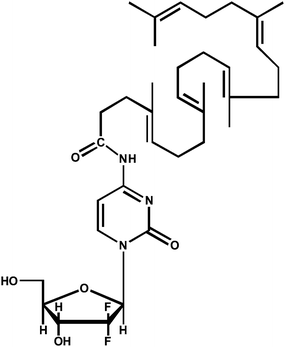 | ||
| Fig. 1 Chemical structure of gemcitabine squalene (GemSQ). | ||
2.3 Preparation of GemSQ liposomes
The DPPC and PEGylated DPPC (DPPC:DSPE-PEG, 95:5) liposomes loaded with GemSQ have been prepared by dissolving phospholipids with GemSQ (15 mol%) in chloroform in order to ensure good homogeneity. Samples were dried under a nitrogen stream and then submitted to low-vacuum freeze drying. The dry films were hydrated with 6 ml of NaCl 0.9% to get a total lipid concentration of 20.0 ± 0.6 mM. The suspensions were heated at 50 °C, above the chain melting transition temperature of DPPC, and vortexed. The resulting MLVs (multilamellar vesicles) were then extruded through a Lipex® extrusion device (Northern Lipids, Canada) under nitrogen pressure. Extrusion was found to be unsuccessful below the gel–fluid transition temperature of phospholipids due to a decrease in membrane fluidity. Therefore, the extruder was thermostated at 50 °C. The extruder was fitted with polycarbonate filters with successive pore size of 0.8 μm, 0.4 μm, 0.2 μm and 0.1 μm as multiple passes through filters were needed to reduce the size of the extruded vesicles.19,20
The hydrodynamic diameters of the resulting liposome suspensions were determined at 25 °C by quasi-elastic light scattering (QELS). The selected scattering angle was 90° (Zetasizer Nano ZS, Malvern Instruments Ltd, UK). The measurements were done 15 min after preparation of the liposomal formulations, in order to have the initial size. The size stability of the suspensions was then investigated over two weeks. The zeta potential of these liposomal formulations was also determined. Each sample was diluted (1/10) in 1 mM NaCl. Analysis of the samples was performed at 25 °C in triplicate.
The final GemSQ concentration in both formulations, GemSQ/DPPC and GemSQ/DPPC/DSPE-PEG, was 2 mg ml−1 for all samples.
2.4 Differential scanning calorimetry
Thermal analysis has been carried out using a DSC 7 (Perkin-Elmer, Inc.) equipped with a cooling device (Intracooler II) in dry air atmosphere. Lauric acid (99.5% purity, melting point Tm = 43.7 °C, enthalpy of ΔHm = 35.713 kJ mol−1) was used as a calibration standard. Thermograms were recorded at 5 °C min−1.Data analysis has been performed using TA Universal Analysis program (New Castle, Delaware, USA). The transition temperatures were taken at the onset of the transitions (Tonset) i.e., the intersection of the tangent to the left side of the endothermic peak with the baseline.
2.5 X-Ray diffraction
X-Ray scattering experiments were performed on the SWING synchrotron beamline at Synchrotron SOLEIL (Gif-sur-Yvette, France). The scattered intensity was reported as a function of the scattering vector q = 4πsinθ/λ, where 2θ is the scattering angle and λ the wavelength of the incident beam. The calibration of the q-range was carried out with pure tristearine (2Lβ form) and silver behenate.2.6 In vivo anticancer activity of liposomal GemSQ formulations
The animal experiments were carried out according to the principles of laboratory animal care and legislation in force in France. DBA/2 mice of 4–5 weeks old weighing approximately 11–15 g were used for this study. The mice were provided with standard mouse food and water ad libitum.The L1210wt leukemia subcutaneous tumor model was developed by injecting the exponentially growing L1210wt leukemia cells in suspension, containing 30% growth factor reduced Matrigel, subcutaneously (1 × 106) into the hind flank region of mice. A palpable tumor was allowed to grow at the injection site. The mice were randomly divided into four groups of eight mice each: untreated, treated with 100 mg kg−1 gemcitabine (MTD), treated with GemSQ/DPPC formulation at 20 mg kg−1 (equivalent in gemcitabine) and treated with GemSQ/DPPC/DSPE-PEG liposomal formulation at 20 mg kg−1 (equivalent in gemcitabine). Six days after tumor implantation, when the mice developed palpable tumors, all groups of mice received the treatment by retro-orbital sinus injection on days 0, 4, 8 and 13, with the exception of the untreated group. The mice were monitored regularly for tumor volume, and survival to assess the anticancer efficacy. All groups were considered statistically valid up to (n − 3) surviving animals. Tumor size was measured across its two perpendicular diameters, and its volume was calculated using the following formula:
| V = 0.5 × (W2 × L) |
2.7 Statistical analysis
Statistical analysis of the in vivo results was performed using Student's t-test considering 95% confidence interval at significance level p < 0.05.3. Results
3.1 Liposome characterization
We have first checked the incorporation of both GemSQ and DSPE-PEG into DPPC bilayers before extrusion, using DSC analysis. As shown in Fig. 2 and Table 1, the pure DPPC bilayers displayed a small pre-transition followed by a sharp main transition at Tm = 42.1 °C corresponding to the gel-to-liquid-crystalline phase transition. Addition of 5 mol% of DSPE-PEG to DPPC modified only slightly the DPPC thermogram. The pre-transition was more diffuse and the onset temperature of the main transition decreases slightly to 41.9 °C. This could be explained by the fine balance between two opposed effects. The main transition temperature of DPPC tended to increase upon mixing with the longer chains of DSPE, whereas lateral repulsive pressure between PEG moieties tended to favour the fluid phase and hence to shift Tm downward. As previously reported, GemSQ and DPPC formed stable homogeneous mixtures in excess water in a wide range of concentrations. In the GemSQ/DPPC mixture (molar ratio r = 0.15) the pre-transition disappeared, a broad and asymmetric transition was observed in the temperature range 32.5–43.5 °C, corresponding to the transition between an inverse bicontinuous cubic phase at room temperature and a fluid lamellar phase at higher temperature.21 Adding 5 mol% of DSPE-PEG induced small changes in DPPC/GemSQ thermograms. The PEGylated formulation exhibited a sharper and more symmetric transition at slightly higher temperature (Tonset = 36.9 °C). Perturbations of DPPC thermal behaviour by GemSQ and DSPE-PEG indicated that these two components were efficiently incorporated into bilayers.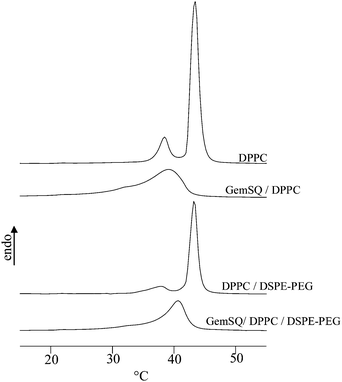 | ||
| Fig. 2 DSC scans of MLV mixtures. Heating cycles were performed at 5 °C min−1. | ||
It is noteworthy that the extrusion of these two highly diluted GemSQ formulations led to the formation of liposomes stable in the 20–50 °C temperature range. The inverse bicontinuous cubic structure of the GemSQ/DPPC mixture was no longer maintained in the nanoassemblies (see below).
Both formulations of liposomes (PEGylated or not PEGylated) obtained by extrusion displayed monodisperse population with diameters close to the average pore size of the last used filter (0.1 μm). Nevertheless, non-PEGylated liposomal formulations exhibited a mean hydrodynamic diameter (133 ± 25 nm, polydispersity index 0.058) tending to be slightly larger than PEGylated liposomes (113 ± 24 nm, polydispersity index 0.035) due to a possible difference in the bilayer curvature.
Both GemSQ/DPPC and GemSQ/DPPC/DSPE-PEG liposomes did not show any visual sedimentation over time. In order to check if aggregation or fusion/coalescence occurred, we have monitored the particle size of the samples during a 15 day period (Table 2). This stability study indicated that the size of the liposomal formulations remained unchanged during this period of evaluation.
The size evolution of GemSQ/DPPC liposomes has also been monitored as a function of temperature. The size of liposomes increased in a reversible manner up to about 145 nm at 50 °C. Interestingly, the ratio between the areas of liposomes at 50 °C and 25 °C (respectively ∼66020 nm2 and ∼53066 nm2) was close to the ratio of the areas per DPPC molecule at these temperatures (respectively 64 Å2 and 47 Å2).22,23 This confirmed that the liposomes formed by extrusion at 50 °C were preserved upon cooling to room temperature.
The zeta potential of the two liposomal formulations has been measured in the presence of 10−3 M NaCl. As expected, GemSQ/DPPC liposomes displayed zeta potential values close to zero. Indeed, DPPC is a zwitterionic phospholipid, globally neutral, and the head-group of GemSQ does not carry charges at neutral pH. On the contrary, the zeta potential of the PEGylated liposomes has been found to be negative (i.e., −35 mV) which may be explained by the net negative charge of the PEG-lipid head-group. This result confirmed that DSPE-PEG was inserted into the bilayer.
The vesicular morphology of the two extruded formulations has been demonstrated by small-angle X-ray scattering (SAXS) measurements (Fig. 3). Regarding the GemSQ/DPPC mixture, the SAXS curve did not show the Bragg reflections indicative of the inverse bicontinuous cubic phase previously evidenced in not extruded GemSQ/DPPC samples at room temperature. A broad bump followed by periodic oscillations was seen in the q-range 0.05–0.5 Å−1. This pattern was characteristic of the bilayer form factor of unilamellar vesicles. From the positions of the curve minima, the thickness of the bilayer could be estimated to be ∼50 Å, consistent with already reported data for pure DPPC. It is noteworthy that the pattern obtained for the PEGylated formulation was the same. The bilayer form factor did not appear to be affected by the hydrophilic PEG layer at the surface of the membrane. This could be explained by the low excess scattering induced by the presence of a small molar fraction of PEG in water.24 Only the electron density profile of the bare bilayer was seen with X-rays. It should be emphasized that the complementary shapes of “inverted wedge-shaped” GemSQ and “wedge-shaped” DSPE-PEG molecules helped to stabilize the vesicles in the PEGylated formulation.
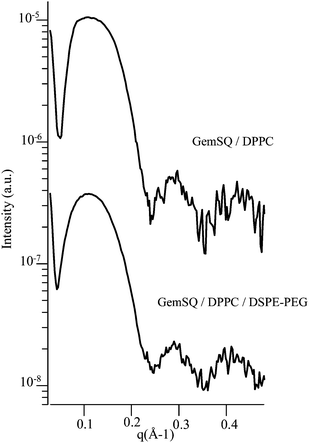 | ||
| Fig. 3 SAXS patterns of GemSQ/DPPC and GemSQ/DPPC/DSPE-PEG extruded liposomes at room temperature (log. scale). | ||
3.2 In vivo anticancer activity of liposomal GemSQ formulations
The anticancer activity of both GemSQ liposomal formulations, (i.e. GemSQ/DPPC and GemSQ/DPPC/DSPE-PEG) was investigated in L1210wt subcutaneously grafted tumors in mice, following intravenous injections on days 0, 4, 8 and 13 (doses of GemSQ, 20 mg, equiv. of gemcitabine). Compared to the untreated control animals, an important inhibition of the tumor growth was observed with similar magnitude whatever the composition of the liposomes (Fig. 4). As a matter of comparison, a comparable antitumor effect was observed with free gemcitabine but at doses 5 to 6.6-times higher than those used for the treatment with GemSQ liposomes (i.e. 100 mg kg−1 for free gemcitabine versus 20 mg kg−1 equiv. gemcitabine for GemSQ). The measure of body weight differences in mice following administration of various formulations did not lead to conclusive results, as the body weights of the treated mice increased progressively due to the growing tumors.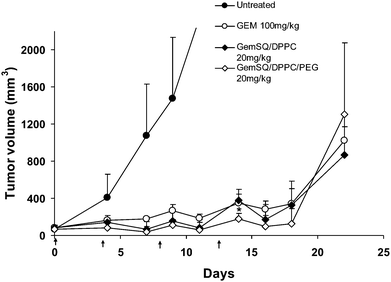 | ||
| Fig. 4 Tumor volume analysis of L1210wt subcutaneous tumor in mice after i.v. injection of gemcitabine (GEM) 100 mg kg−1 and liposome formulations of gemcitabine squalene (GemSQ) 20 mg kg−1 eq. of gemcitabine. A palpable tumor was developed on the hind flank region of mice by subcutaneously injecting 1 × 106 L1210wt cells in suspension containing 30% growth factor reduced Matrigel. The above formulations were injected intravenously into the tumor-bearing mice on days 0, 4, 8, 13, indicated by ↑ in the figure. All groups were considered statistically valid up to (n − 3) surviving animals. The results are expressed as mean ± S.D. Statistical analysis was performed using Student's t-test considering 95% confidence interval at significance level p < 0.05 (* indicates p < 0.05). | ||
4. Discussion
Liposomes are generally considered as non-toxic nanovesicles even when administered at very high dose and this makes them attractive as drug delivery vehicles.25 Although not all hydrophilic and lipophilic molecules could be entrapped into liposomes with sufficient drug loading, in some cases ionic interactions with the lipids may allow satisfying entrapment efficiencies,26 while in few cases specific lipid composition may also improve the encapsulation.27 On the other hand, long circulating liposomes obtained by surface coating with polyethyleneglycol may provide many advantages in cancer targeting since they can accumulate in tumors, due to their ability to extravasate into these tissues by enhanced permeability and retention effect. However, the design of long circulating liposomes with encapsulated hydrophilic compounds, like gemcitabine, is a real challenge since the drug release may occur during the residence time of the liposomes in the blood circulation, before accumulation into the tumor tissue.In general, the lipophilic prodrug approach facilitates the incorporation of both hydrophilic and hydrophobic small therapeutic molecules into liposomal vesicles.28 For this reason, the design of an altered lipophilic prodrug of gemcitabine that would be more efficiently retained in liposomes is an interesting alternative approach. Thus, we have employed this approach in the present study by coupling gemcitabine with squalene, a natural lipid and a precursor in the biosynthesis of cholesterol. It was expected that the stable anchoring of this lipophilic bioconjugate in the liposomal bilayer could lead to significant anticancer activity in experimental tumors.
The incorporation of GemSQ into DPPC and DPPC/DSPE-PEG bilayers has been evidenced by DSC analysis, whereas the vesicular morphology of both formulations has been demonstrated by SAXS measurements. The prepared GemSQ/DPPC and GemSQ/DPPC/DSPE-PEG formulations were found to be stable for at least 2 weeks allowing us to perform in vivo experiments in good conditions; the size of the liposomes (i.e. 113 nm and 130 nm) were compatible with intravenous injection.
We show in this paper that the liposomal formulations of GemSQ displayed at doses as low as 20 mg kg−1 (equiv. of gemcitabine) (the maximum tolerable dose of GemSQ) a similar anticancer activity to free gemcitabine injected at a dose of 100 mg kg−1. However, contrary to what was expected, the PEG-coated liposomes did not improve considerably the antitumoral effect as compared with plain, non-PEGylated liposomes, although the antitumor activity difference of the PEGylated liposomal formulation over the non-PEGylated liposomal formulation was statistically significant at a single time point on day 14. Vascularization and microvessels density have, however, been previously evidenced by immunohistochemistry in the leukemia solid tumor model chosen for this study. It is noteworthy that the subcutaneous implanted version of the L1210wt leukemia has already been employed to study the tumor penetration of fluorescent latex particles mimicking liposomes29 and to determine the antitumor activity of antisense oligonucleotide loaded into lipid nanoparticles against the anti-apoptotic Bcl2.30 In our case, it remains, however, possible that the subcutaneously growing L1210wt leukemia did not display the increased permeability of tumor vessels, needed for the “enhanced permeability and retention” (EPR) effect31 to occur, possibly attributed to the smaller initial tumor size as compared to that reported in Pan et al.29 This may perhaps be one of the reasons why the PEGylated GemSQ liposomes did not display superior anticancer activity compared to their non-PEGylated counterpart. It might also be speculated that a fraction of GemSQ was transferred from liposomes to lipoproteins in the blood stream. Indeed, this effect has already been reported for annamycin, a lipophilic derivative of doxorubicin.32 When liposomes containing annamycin were incubated in plasma, the major part of annamycin was recovered in the HDL fraction of plasma lipoproteins. “Stealth” liposomes did not display any advantage concerning the tumor targeting compared to bare liposomes. It is likely that they did not significantly prevent the transfer of annamycin to blood components.33 The same effect was observed for a lipophilic derivative of Ara-C, NHAC. The drug was rapidly transferred from liposomes to plasma proteins and erythrocytes, regardless of the liposome’s surface coating with PEG.11 Dosages of GemSQ in LDL, HDL and/or VLDL should allow one to clarify if an exchange of the prodrug may exist between liposomes and lipoproteins. If so, a modification of the lipid composition of the liposomes should be considered in order to reduce GemSQ exchange with lipoproteins. The body weight differences in mice following the treatments were assessed to understand if the PEGylated liposomal formulation could make a difference over non-PEGylated formulation in terms of toxicity; however no meaningful conclusion could be made from these observations. This is because the L1210wt subcutaneous tumor chosen in this study is a rapidly growing tumor and hence the body weights of the tumor-bearing mice increased faster than the decrease in body weights, if any, caused by the treatment.
Thus, in the tumor model tested in our study, it may be hypothesized that the anticancer efficacy of the liposomal formulations at considerably low doses compared to the free drug was principally due to the protection of gemcitabine from deamination in the blood stream since the PEGylation did not lead to a considerable advantage compared to non-PEGylated liposomes.
5. Conclusion
In this manuscript, an attempt has been made to develop liposomal formulations of gemcitabine by using a lipidic prodrug of this compound, i.e. GemSQ, in order to facilitate the encapsulation. The incorporation of this prodrug into the liposomal bilayers has been experimentally confirmed employing DSC and SAXS. Contrary to what was expected, the PEGylated liposomal formulation did not, however, exhibit superior anticancer activity over the non-PEGylated liposomal formulation in the tumor model chosen. Further in vivo follow-up studies such as pharmacokinetic profiling and activity studies in other relevant preclinical models would facilitate a better understanding of these observations.References
- R. Colomer, Oncology (Williston Park), 2004, 18, 8 Search PubMed.
- R. J. Schilder, J. A. Blessing, M. Morgan, C. E. Mangan and J. S. Rader, Gynecol. Oncol., 2000, 76, 204 CrossRef CAS.
- L. Toschi, G. Finocchiaro, S. Bartolini, V. Gioia and F. Cappuzzo, Future Oncol., 2005, 1, 7 Search PubMed.
- A. Hilbig and H. Oettle, Expert Rev. Anticancer Ther., 2008, 8, 511 Search PubMed.
- L. H. Reddy and P. Couvreur, Curr. Pharm. Des., 2008, 14, 1124 CrossRef CAS.
- M. Celano, M. G. Calvagno, S. Bulotta, D. Paolino, F. Arturi, D. Rotiroti, S. Filetti, M. Fresta and D. Russo, BMC Cancer, 2004, 4, 63 CrossRef.
- R. Moog, A. M. Burger, M. Brandl, J. Schuler, R. Schubert, C. Unger, H. H. Fiebig and U. Massing, Cancer Chemother. Pharmacol., 2002, 49, 356 CrossRef CAS.
- C. Celia, M. G. Calvagno, D. Paolino, S. Bulotta, C. A. Ventura, D. Russo and M. Fresta, J. Nanosci. Nanotechnol., 2008, 8, 2102 CrossRef CAS.
- D. Cosco, A. Bulotta, M. Ventura, C. Celia, T. Calimeri, G. Perri, D. Paolino, N. Costa, P. Neri, P. Tagliaferri, P. Tassone and M. Fresta, Cancer Chemother. Pharmacol., 2009, 64, 1009 CrossRef CAS.
- M. G. Calvagno, C. Celia, D. Paolino, D. Cosco, M. Iannone, F. Castelli, P. Doldo and M. Fresta, Curr. Drug Delivery, 2007, 4, 89 Search PubMed.
- R. Schwendener and H. Schott, Methods Enzymol., 2005, 391, 58 CAS.
- M. L. Immordino, P. Brusa, F. Rocco, S. Arpicco, M. Ceruti and L. Cattel, J. Controlled Release, 2004, 100, 331 CrossRef CAS.
- P. Brusa, M. L. Immordino, F. Rocco and L. Cattel, Anticancer Res., 2007, 27, 195 CAS.
- P. Couvreur, L. H. Reddy, S. Mangenot, J. H. Poupaert, D. Desmaële, S. Lepêtre-Mouelhi, B. Pili, C. Bourgaux, H. Amenitsch and M. Ollivon, Small, 2008, 4, 247 CrossRef CAS.
- P. Couvreur, B. Stella, L. Harivardhan Reddy, H. Hillaireau, C. Dubernet, D. Desmaële, S. Lepêtre-Mouelhi, F. Rocco, N. Dereuddre-Bosquet, P. Clayette, V. Rosilio, V. Marsaud, J. M. Renoir and L. Cattel, Nano Lett., 2006, 6, 2544 CrossRef CAS.
- L. H. Reddy, P. E. Marque, C. Dubernet, S. Lepêtre-Mouelhi, D. Desmaële and P. Couvreur, J. Pharmacol. Exp. Ther., 2008, 325, 484 CrossRef CAS.
- L. H. Reddy, C. Dubernet, S. Lepêtre-Mouelhi, P. E. Marque, D. Desmaële and P. Couvreur, J. Controlled Release, 2007, 124, 20 CrossRef CAS.
- L. H. Reddy, H. Khoury, A. Paci, A. Deroussent, H. Ferreira, C. Dubernet, X. Declèves, M. Besnard, H. Chacun, S. Lepêtre-Mouelhi, D. Desmaële, B. Rousseau, C. Laugier, J.-C. Cintrat, G. Vassal and P. Couvreur, Drug Metab. Dispos., 2008, 36, 1570 CrossRef CAS.
- P. J. Patty and B. J. Frisken, Biophys. J., 2003, 85, 996 CrossRef CAS.
- B. J. Frisken, C. Asman and P. J. Patty, Langmuir, 2000, 16, 928 CrossRef CAS.
- B. Pili, C. Bourgaux, H. Amenitsch, S. Lepêtre-Mouelhi, D. Desmaële, P. Couvreur and M. Ollivon, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1522 CrossRef CAS.
- N. Kucerka, S. Tristram-Nagle and J. F. Nagle, Biophys. J., 2006, 90, L83 CrossRef CAS.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, 2000, 1469, 159 CrossRef CAS.
- L. Arleth, B. Ashok, H. Onyuksel, P. Thiyagarajan, J. Jacob and R. P. Hjelm, Langmuir, 2005, 21, 3279 CrossRef CAS.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145 CrossRef CAS.
- V. Joguparthi and B. D. Anderson, J. Pharm. Sci., 2008, 97, 433 CrossRef CAS.
- S. Koudelka, P. Turanek-Knotigova, J. Masek, Z. Korvasova, M. Skrabalova, J. Plockova, E. Bartheldyova and J. Turanek, J. Pharm. Sci., 2010, 99, 2309 CAS.
- R. A. Schwendener and H. Schott, Methods Mol. Biol., 2010, 605, 129.
- X. Pan, R. J. Lee and M. Ratnam, Anticancer Res., 2004, 24, 3005.
- X. Pan, L. Chen, S. Liu, X. Yang, J. X. Gao and R. J. Lee, Mol. Pharmaceutics, 2009, 6, 211 CrossRef CAS.
- H. Maeda, T. Sawa and T. Konno, J. Controlled Release, 2001, 74, 47 CrossRef CAS.
- Y. Zou, Y. H. Ling, S. Reddy, W. Priebe and R. Perez-Soler, Int. J. Cancer, 1995, 61, 666 CrossRef CAS.
- K. M. Wasan and R. E. Morton, Pharm. Res., 1996, 13, 462 CrossRef CAS.
Footnote |
| † Current address: Sanofi-aventis, 13 Quai Jules-Guesdes, 94403, Vitry-sur-Seine, France. |
| This journal is © The Royal Society of Chemistry 2010 |