Intracellular trafficking of silicon particles and logic-embedded vectors†
Silvia
Ferrati
a,
Aaron
Mack
a,
Ciro
Chiappini
b,
Xuewu
Liu
a,
Andrew J.
Bean
cd,
Mauro
Ferrari‡
aef and
Rita E.
Serda‡
*a
aUniversity of Texas Health Science Center, Department of NanoMedicine and Biomedical Engineering, 1825 Pressler Street, Suite 537, Houston, TX 77030, USA. E-mail: rita.serda@uth.tmc.edu; Tel: +713-500-2309
bThe University of Texas at Austin, Austin, TX 78712, USA
cUniversity of Texas Health Science Center, Department of Neurobiology and Anatomy, Houston, TX 77030, USA
dUniversity of Texas MD Anderson Cancer Center, Department of Pediatrics, Houston, TX 77030, USA
eUniversity of Texas MD Anderson Cancer Center, Department of Experimental Therapeutics, Houston, TX 77030, USA
fRice University, Department of Bioengineering, Houston, TX 77005, USA
First published on 7th June 2010
Abstract
Mesoporous silicon particles show great promise for use in drug delivery and imaging applications as carriers for second-stage nanoparticles and higher order particles or therapeutics. Modulation of particle geometry, surface chemistry, and porosity allows silicon particles to be optimized for specific applications such as vascular targeting and avoidance of biological barriers commonly found between the site of drug injection and the final destination. In this study, the intracellular trafficking of unloaded carrier silicon particles and carrier particles loaded with secondary iron oxide nanoparticles was investigated. Following cellular uptake, membrane-encapsulated silicon particles migrated to the perinuclear region of the cell by a microtubule-driven mechanism. Surface charge, shape (spherical and hemispherical) and size (1.6 and 3.2 μm) of the particle did not alter the rate of migration. Maturation of the phagosome was associated with an increase in acidity and acquisition of markers of late endosomes and lysosomes. Cellular uptake of iron oxide nanoparticle-loaded silicon particles resulted in sorting of the particles and trafficking to unique destinations. The silicon carriers remained localized in phagosomes, while the second stage iron oxide nanoparticles were sorted into multi-vesicular bodies that dissociated from the phagosome into novel membrane-bound compartments. Release of iron from the cells may represent exocytosis of iron oxide nanoparticle-loaded vesicles. These results reinforce the concept of multi-functional nanocarriers, in which different particles are able to perform specific tasks, in order to deliver single- or multi-component payloads to specific sub-cellular compartments.
Introduction
Logic-embedded vectors (LEVs),1 used for delivery of therapeutic or imaging agents, are designed to perform time-sequences of actions in order to overcome biological barriers, reach targeted lesions, and deliver single- or multi-component payloads, which may be directed to localize in specific sub-cellular compartments.2,3 We have fabricated multi-functional nanocarriers (i.e. a multi-stage delivery system) for disease targeting. These nanocarriers consist of mesoporous silicon particles that are discoidal and hemispherical in shape and serve as first-stage particles in multi-particle constructs. The multi-stage system is assembled by loading secondary nanoparticles (second-stage particles) into the pores of the silicon particles.4Loading nanoparticles into porous silicon matrices provides tuning at multiple levels to achieve diverse functions. Tunable parameters include particle geometry and surface modification, as well as tunable rates of degradation based on silicon porosity.5,6 Moreover, factors impacting cellular internalization and trafficking of the first-stage silicon particle also influence the release location and biological impact of the second-stage particles. Additionally, the intrinsic nature of the second-stage particle plays a large role in its intracellular trafficking and final subcellular localization, providing a mechanism to redirect nanoparticles to individual organelles or to make them responsive to local stimuli, such as pH changes.7 Despite significant advances in studying the behavior of these carriers in biological systems8–10 a complete understanding of the intracellular trafficking of LEVs is still not available.
In the present study we have examined the intracellular trafficking of first-stage silicon carriers and multi-stage LEVs. The LEV was assembled by loading superparamagnetic iron oxide nanoparticles (SPIONs), coated with amino-PEG, into porous silicon particles. The goal of this project was to define the cellular trafficking and transport of LEVs using confocal microscopy combined with ultrastructural examination to provide an overview of the spatio-temporal characteristics of particle trafficking, such as mobility and trajectories in real time, as well as cellular localization of particles. Since vascular endothelial cells have been shown to be possible candidates for nanocarrier targeting, and previous results show their ability to act as non-professional phagocytes capable of internalizing silicon microparticles,9 we chose human microvascular vein endothelial cells (HMVECs) as the cellular model for our study.
Results and discussion
Silicon particle uptake by human microvascular vein endothelial cells
Herein we elucidate the intracellular trafficking of our nanodelivery platform, consisting of SPIONs (15 nm) encapsulated within the pores of silicon particles. The trafficking of the silicon particles was studied prior to assembling the multi-stage system to investigate the fate of the first-stage particles. In general, particles smaller than 500 nm are internalized through receptor-mediated endocytosis while larger particles are internalized through phagocytosis. We have previously shown that nanoporous silicon carriers are internalized by endothelial cells through a combination of phagocytosis and macropinocytosis.9,11 Both mechanisms are actin-driven processes that involve extensive membrane re-organization with the formation of pseudopodia and extension of the cell membrane to surround and engulf the particles. Since the biological impact of nanocarriers depends not only on cellular uptake, but on the subcellular location after internalization, we examined the intracellular destination of silicon particles by transmission electron microscopy (TEM). Since charge and size can potentially affect the subcellular localization of particles, four different types of silicon particles were studied: two sizes (1.6 μm and 3.2 μm), each presenting positive and negative charges. HMVECs were incubated with silicon particles for 6 h; the cells were then fixed, and processed for TEM analysis. In Fig. 1A, positively charged particles of both sizes are displayed. Membranes can be seen surrounding each type of particle, predominately as a tight-fitting enclosure. Internalized negative particles are shown in Fig. S1 of the ESI.† Membrane entrapment appeared similar for both particle sizes, independent of surface charge. Intracellular localization of microparticles is shown after 6 h to allow for delayed uptake of negative, oxidized microparticles. As reported previously, endothelial cells preferentially internalize positive microparticles in the presence of serum, and in this study uptake of negative microparticles was both reduced and delayed. Cellular uptake at later time points may reflect degradation of the silicon surface and thereby altered binding by serum components.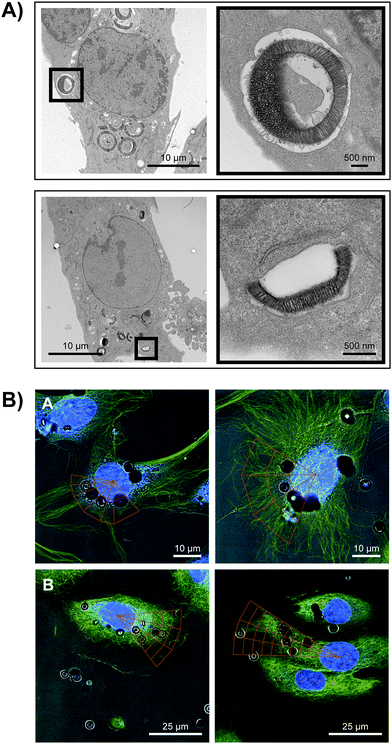 | ||
| Fig. 1 Perinuclear trafficking of hemispherical silicon particles. (A) TEM micrographs of APTES-modified silicon particles 6 h after introduction to HMVECs (top: 3.2 μm; bottom: 1.6 μm particles). (B) Confocal images of particles internalized in non-treated control (top row) or nocodazole-treated (150 nM; bottom row) HMVECs visualized with DRAQ5 (nuclei) and FITC-conjugated α-tubulin antibody, 5 h after particle addition. The localization of particles with respect to the nucleus was quantified by dividing portions of cells in quadrants, having defined distances from the nucleus. | ||
Microtubule-mediated transport
Following cellular uptake, phagosome maturation is promoted by a microtubule-mediated system that propels vesicles containing the particles towards the perinuclear region of the cell. A number of proteins participate at various stages in the endocytic pathway by associating transiently with the maturing endo/phagosomes. For example, EEA1 and Rab 5 associate with newly formed compartments (early endo/phagosomes) while Lamp1 and NPC1 associate with later compartments (late endosomes and lysosomes) in the pathway.12 These proteins promote the association of endo/phagosomes with microtubules and motor proteins, originating the centripetal movement of the vesicle.13 Maturation of the phagosome is inhibited by microtubule disruption, supporting the link between phagosome maturation and microtubule-mediated transport.14–16 To determine whether the trafficking mechanism for the movement of encapusulated microparticles is dependent on classical trafficking machinery, we incubated HMVECs with 3.2 μm silicon particles in the presence or absence of 150 nM nocodazole, a drug that alters microtubules dynamics. Accumulation of particles in the perinuclear region of cells was determined by confocal microscopy using DRAQ5 and FITC-conjugated antibodies specific for nuclear and α-tubulin staining respectively (Fig. 1B). The subcellular distribution of particles was quantified by dividing the cells into quadrants having defined distances from the nucleus and counting the particles in each quadrant. The alteration of the microtubule network affected the motility of individual microparticle-laden phagosomes, reducing their accumulation in the perinuclear region by 80%. These data suggest that microtubule-based movement of phagosomes is required for redistribution of silicon particles within the cell.Maturation of phagosomes bearing silicon particles
During phagosome maturation, the phagosome undergoes a gradual but consistent acidification from pH 7 to 5 due to the function of a membrane ATPase acquired by fusion of the phagosome with vesicles containing acidic compartments. Maximal acidification is achieved upon fusion of the phagosome with lysosomes.16 To verify that the presence of the microparticles does not perturb the regular intracellular pathways of maturation, and to obtain information about the fate of the particles over longer time scales, 3.2 μm discoidal particles were conjugated with a pH-sensitive dye (pH Rhodo; Invitrogen) that becomes fluorescent in an acidic environment. Prior to the experiment, the utility of the particles was tested in progressively more acidic solutions and an increase in fluorescence intensity was verified from a value of 2.2 at pH 7 to a value of 27.0 at pH 4. HMVECs were then incubated with the modified microparticles for varying amounts of time and analyzed using flow cytometry. Side scatter and fluorescence intensity were used as metrics to monitor cellular uptake and maturation of the vesicle, making it possible to follow the fate of the carriers inside of the cells over time.10 As shown in Fig. 2A, the mean fluorescent intensity of cells incubated with particles increased progressively with longer incubation times, from an average intensity near 5 at time zero, when particles are mostly outside the cells, to an intensity of 19 after 16 h of incubation, when particles are present in compartments that have undergone a progressive decrease in pH. There is a slight decrease in fluorescence following 24 h of incubation that may reflect degradation of the particle surface and partial release of the pH Rhodo dye. The overall trend indicates that after internalization of the particles into phagosomes, the compartments mature and the environment surrounding the particle becomes increasingly more acidic. Expressing GFP-tagged proteins that localize in a compartment-specific manner and then co-localizing the internalized particles with these markers is another method for determining the localization of the particles. HMVECs were transfected with GFP fused to Niemann Pick C1 (NPC1), a lysosomally localized protein, and then incubated with Dylight 594-labelled 3.2 μm silicon particles for 4 h. In Fig. 2B, confocal images are presented as separate or merged channels. GFP-NPC1 was expressed in regions surrounding some of the internalized particles, especially those localized in the perinuclear region of the cell, and could be observed to form green rings around the particles. These data suggest that the particles were localized in the lysosome. Since any modification of the particle, including attachment of a fluorophore, could potentially alter the intrinsic distribution of the particles, we repeated the experiment using unlabeled particles and bright-field imaging to visualize the particles (Fig. 2B). Similar results were obtained.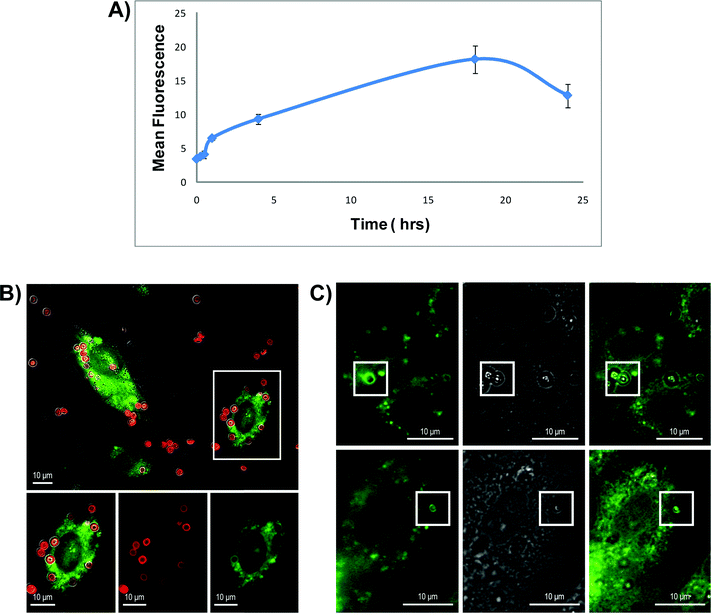 | ||
| Fig. 2 Colocalization of silicon particles with NPC1 protein and acidic vesicles. (A) HMVECs were incubated with 3.2 μm discoidal silicon particles conjugated with a pH-sensitive dye (pH Rhodo). Mean fluorescence was monitored over 24 h by flow cytometry. (B)–(C) Confocal images of GFP-NPC1 transfected HMVECs incubated 4 h with Dylight 594-labelled (B) or un-labelled (C) silicon particles. The images are reported in separated and merged channels. | ||
Intracellular mobility and trajectory of the silicon particles
In order to further characterize the intracellular transport and mobility of our nanocarriers over various sizes, shapes and surface charges, we quantified the trajectory, directionality, and rate of intracellular migration of silicon particles using real-time confocal microscopy. Images were taken in 5 focal planes every 5 min and movies were compiled (see the ESI†). In addition to our four variations of porous silicon microparticles, three sizes of spherical silica beads (1, 2.5, and 3 μm) were included to further study size effects. As shown in Fig. 3A, after cellular uptake particles migrated from the cellular membrane towards the perinuclear region, leading to their accumulation in that region. Manual tracking of the particles inside the cells was performed using the Slide Book software. The particles were tracked from the moment they reached the cell surface until they reached the perinuclear region and the entire process for each particle took about 50 min. From the two-dimensional time dependent coordinates [x(t), y(t)] of the particles individual time-averaged mean square displacements (MSDs), 〈Δr2(τ)〉, were calculated according to the following equation:| 〈Δr2(τ)〉 = 〈[x(t + τ) − x(t)]2 + [y(t + τ) − y(τ)]2〉 |
![Effect of size and shape on the rate of particle migration. (A) Manual tracking from the point of uptake to perinuclear region was done by confocal imaging of cells every 5 min (bar 10μm). (B) (top) An example of MDS curve fitting and (bottom) box charts showing the calculated rates of migration toward the perinuclear region for different types of particles [‘Ox’ and ‘APTES’ are modified hemispherical silicon particles (two sizes), and ‘Beads’ are spherical silica particles (three sizes)]. Populations were not significantly different based on ANOVA test with P = 0.05.](/image/article/2010/NR/c0nr00227e/c0nr00227e-f3.gif) | ||
| Fig. 3 Effect of size and shape on the rate of particle migration. (A) Manual tracking from the point of uptake to perinuclear region was done by confocal imaging of cells every 5 min (bar 10μm). (B) (top) An example of MDS curve fitting and (bottom) box charts showing the calculated rates of migration toward the perinuclear region for different types of particles [‘Ox’ and ‘APTES’ are modified hemispherical silicon particles (two sizes), and ‘Beads’ are spherical silica particles (three sizes)]. Populations were not significantly different based on ANOVA test with P = 0.05. | ||
To analyze the trajectories the MSD curves were first fitted with the general equation: y = 6Dtk(fitting 1). If k = 1 transport follows Fick's law, which describes random thermally driven motion. When k >1 active transport is indicated, and particles migrate in a given direction. Since all our curves were fit using an exponent larger than 1, we analyzed movement based on active transport. The transport rates of carriers exhibiting active transport are described by 〈Δr2(τ)〉 = 4Dt + υ2t2 (fitting 2) where the first term is the random diffusion contribution, where D is the diffusion coefficient, and the second term is the active transport contribution caused by directed motion, where υ is the rate of intracellular trafficking of the carrier.17 As shown in Fig. 3B, from the fitting of the MSDs, we calculated the rate of transport for each type of microparticle, obtaining an average rate of trafficking toward the perinuclear region of 0.5 μm per minute for all particle populations. An ANOVA test (P = 0.05) was used to compare populations. Thus particle size (1–3.2 μm), surface charge (positive or negative), and shape (hemispherical or spherical) did not impact the rate of vesicle transport. The data, as well as the perinuclear localization of the phagosome-containing particles suggests that the particle-loaded phagosomes are able to mature and that the presence of the particles does not arrest the movement or localization of the endo/phagosomes.
Assembly and characterization of LEVs
LEVs were assembled using 3.2 μm oxidized discoidal silicon particles with mean pore size of 50 nm and 15 nm SPIONs coated with amino-PEG. TEM images of the loaded silicon particles are shown in Fig. 4D. The SPIONs were selected based on earlier findings that indicated endosomal sorting of amino-PEG-coated SPIONs from carrier silicon particles.18 Specific surface including polyethyleneimine (PEI),19 poly(lactic acid-co-glycolic acid) (PLGA)20 and chitosan.21,22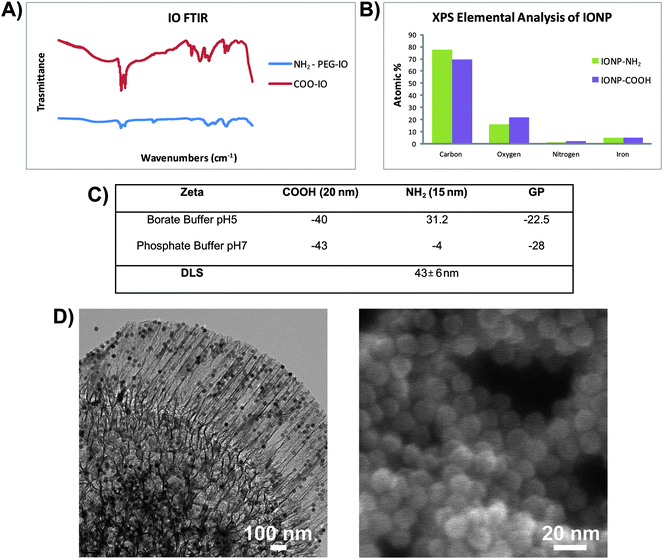 | ||
| Fig. 4 Particle characterization and loading FTIR spectra (A) and XPS elemental analysis (B) of carboxylated (IONP-COOH) and amine-PEG (IONP-NH2) SPIONs. (C) Zeta-potential and dynamic light scattering (DLS) measurements for SPIONs and silicon particles in phosphate and borate buffers. (D) TEM (left) and SEM (right) micrographs showing 3.2 μm discoidal particles loaded with amino-PEG SPIONs. | ||
SPIONs were characterized using Fourier transform infrared (FTIR) spectroscopy and zeta-potential analysis. FTIR spectra were acquired for amino-PEG SPIONs and compared with carboxylated SPIONs (Fig. 4A). Carboxylated particles presented four characteristic bands: 3189 cm−1 (O–H), 1702 cm−1 (COOH) and two bands around 2922 cm−1 and 2852 cm−1 (C–H stretching). Amine-PEG also present bands at 2922–2853 cm−1 (CH stretching) in addition to the characteristic primary amine bands at 3341 cm−1 (NH) and 1298 cm−1 (CN). They also display the PEG bands: 1700 cm−1 (COOH) and 1104 cm−1 (CH2–OH) and the ammonium peak (NH4+) around 1418 cm−1. Furthermore, XPS elemental analysis of particles confirmed a decrease in the oxygen peak when comparing carboxylated SPIONs (COOH-SPIONs) to the amine-PEG SPIONs. (Fig. 4B).
The zeta-potential, which is indicative of the net surface charge, was determined for each particle. Measurements were taken both in phosphate buffer (pH 7) and borate buffer (pH 5), the latter being the loading buffer for particle assembly. Silicon particles and carboxylated SPIONs were negatively charged in both buffers while amine-PEG SPIONs were neutral and positively charged in phosphate and borate buffer respectively (Fig. 4C). The stability of the polymer coating on the SPIONs was studied by incubating the particles in phosphate buffer pH 7 for 6 days and checking for surface charge repeatedly. The surface charge remained unchanged over the period of analysis indicating stability of the surface. DLS measurements of the SPIONs in phosphate buffer were also performed to provide the hydrodynamic radius of the particles (Table C in Fig. 4).
Mesoporous silicon particles (oxidized, negative charge) were dried and loaded with the amine-PEG SPIONs by capillary action and electrostatic interactions (Fig. 4D). The loading efficiency was quantified by inductively coupled plasma optical emission spectrometry (ICP-OES) with a resulting efficiency of 21% (equivalent to 6.4 μg per 3 × 106 silicon particles).
Intracellular trafficking of LEVs and exocytosis of SPIONs
LEVs were incubated with HMVECs and the intracellular location of particles was determined by TEM. At 24 and 32 h, the nanoparticles were predominantly associated with the silicon particles, and encapsulated in the same phagosome. In some endosomes, regions rich in SPIONs started to bud from the primary vesicle as shown in Fig. 5, leading to the formation of novel compartments containing multivesicular bodies. This suggests that the SPIONs are actively sorted from the original endosomes into unique vesicles that are then free to independently traffic in the cell.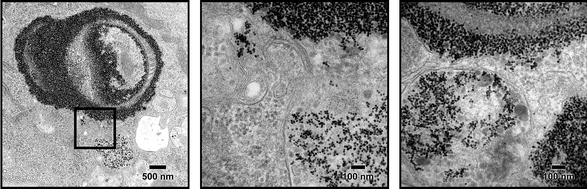 | ||
| Fig. 5 TEM micrographs of silicon particles loaded with amine-PEG SPIONs 24 h after internalization by HMVECs showing endosomal sorting of SPIONs into multi-vesicular bodies (MVB) and the process of MVB budding into separate membrane-bound compartments. | ||
After observing that second-stage nanoparticles move to vesicles separated from the silicon carriers, and based on reports of exocytosis of nanoparticles from eukaryotic cells,18,23 we examined the supernatant of cells incubated with LEVs at multiple time points within a 7 day period for released SPIONs. The amount of iron released was quantified using ICP-OES and normalized for the iron present in the media of untreated cells. The data were expressed as percentage of iron released with respect to iron effectively delivered to cells [amount initially incubated with cells minus that removed in the media at 12 h (i.e. SPIONs not internalized)]. The average amount of SPIONs in the supernatant, tested in unique cultures on different days was 25%. A TEM image showing a cell containing an empty silicon particle after 7 days of incubation with cells supports the process of release of SPIONs from the carrier silicon particle with the concurrent endosomal retention of the carrier particle (Fig. 6A). These results were compared with cells treated with free SPIONs in which the release of SPIONs into the media was 16% (Fig. 6B). The higher release of SPIONs from cells incubated with LEVs may result from coordinated endosomal sorting and cellular release or from differences in the absolute amount of SPIONs per cell and its impact on cellular secretion.
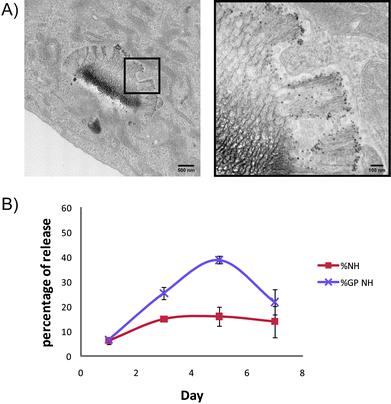 | ||
| Fig. 6 Endosomal sorting and secretion of SPIONs (A) TEM images showing a HMVEC containing a membrane-bound silicon particle with the majority of SPIONs released 7 days after LEV introduction. (B) SPIONs secretion from cells over 7 days following treatment with free or silicon particle-delivered SPIONs (‘GP’ is silicon particles, ‘NH’ is amine-PEG coated SPIONs). | ||
Conclusions
In summary we have elucidated the cellular trafficking of porous silicon carriers and LEVs, consisting of silicon carriers loaded with amine-PEG functionalized SPIONs, in endothelial cells. We have shown that the silicon microparticles, regardless of size and surface functionalization, were internalized through phagocytosis resulting in entrapment in phagosomes. The phagosomes were able to mature and traffic using an active transport process that is consistent with microtubule-based movement toward the perinuclear region. Trafficking of silicon carriers was characterized in terms of rate of intracellular migration, and no impact of microparticle charge or size was observed. Following cellular uptake of the LEV, SPIONs encapsulated within the pores of the silicon particles were released from the silicon particle and sorted into MVBs. The MVBs formed unique vesicles that appear to be candidates for cellular secretion. TEM images showing extracellular vesicles containing SPIONs have been reported in macrophages that were incubated with the LEV.25 Secretion of cell-derived vectors containing nanoparticles can potentially redirect selected payloads to neighboring cells and may represent an important mechanism for cell-to-cell communication.Material and methods
Porous silicon particles and their surface modification
Nanoporous silicon microparticles were fabricated by our group using semiconductor microfabrication and electrochemical etching in the Microelectronics Research Center at the University of Texas at Austin.24 The mean particle diameter for hemispherical particles was either 1.6 ± 0.2 or 3.2 ± 0.2 μm, with a pore size of 26.3 ± 14.6 μm. Discoidal particles had a diameter of 3.2 ± 0.2 μm with average pore size of 51.3 nm. Silicon microparticles were oxidized with piranha solution, creating a negative surface charge and functional groups for further modification, with 3-aminopropyl-triethoxysilane (APTES) providing a positive surface charge. The details of the surface modification were recently published.8Dynamic light scattering (DLS) and zeta-potential analysis
Amine-PEG coated superparamagnetic iron oxide nanoparticles (15 nm, SPIONs) were purchased from Ocean NanoTech (Springdale Arkansas). Size and charge measurements were performed using a Zeta PALS Zeta Potential Analyzer equipped with 90Plus/BI-MAS Multi Angle Particle Sizing Option (Brookhaven Instruments Corporation; Holtsville, NY). Charge measurements were taken in both phosphate buffer (pH 7) and borate buffer (pH 5). The refractive index was set at 1.4 and the dust cut-off filter was set at 50. Each final value is the average of four measurements. DLS samples were measured at 90° at 25 °C. To test the stability of the surface coatings, repeated zeta-potential measurements were performed over 6 days in phosphate buffer.X-Ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR)
XPS elemental analysis and FTIR were performed to qualitatively show the difference in the particle coatings. Samples for XPS were prepared by applying 20 μl of 1 mg mL−1 amino-PEG coated (SPIONs-NH2) and carboxylated (SPIONs-COOH) iron oxide nanoparticles to a silicon stub and dried overnight in a dessicator. Measurements were performed on a PHI Quantera XPS over the range of 1100–0 eV. Elemental peaks were identified and analyzed with PHI's Multipak software. Samples for FTIR were prepared applying them to the diamond surface of a SMART ATR attachment on a Nicolet 6600 FTIR spectrophotometer. The samples were then dried with nitrogen and the spectra read. The room-temperature detector was used to collect all data and all readings were made using a resolution of 4 cm−1 and by averaging 16 readings in absorbance mode in order to form the final curve. Analysis of the peaks was performed using Omnic peak identification software.Porous silicon nanocarrier loading
Oxidized discoidal silicon particles (1 × 107, 3.2 μm) were collected by centrifugation in IPA and dried overnight at room temperature in vacuum desiccators. 100 μg of the 15 nm amino-PEG coated SPIONs in borate buffer at 1 mg ml−1 were added to the silicon particles for loading. The particle suspension was sonicated and incubated for 30 min at room temperature. Samples were then centrifuged at 2000 rpm (Beckman Coulter Allegra X-22 Centrifuge equipped with a 296/06 rotor) to remove the free unloaded nanoparticles, followed by two washes with water. Loaded silicon particles were resuspended in medium and incubated with cells. The loading efficiency was obtained by measuring iron content in the supernatant and the particle pellet using inductively coupled plasma optical emission spectrometry (ICP-OES). The instrument was a Varian Vista AX at a power of 1 kW, with plasma flow set to 15 L min−1, auxiliary flow of 1.5 L min−1 and a nebulizer flow of 0.75 L min−1, with 5 replicate readings at 15 s between each reading.Cell culture
Human microvascular vein endothelial cells (HMVECs) were a kind gift from Dr Rong Shao at the University of Massachusetts. Cells were cultured as a monolayer in EBM medium (Clonetics, Walkersville, MD USA). They were maintained at 37 °C in a humidified 5% CO2 atmosphere and detached using 0.25% mg ml−1 trypsin/EDTA solution (Clonetics) upon reaching 80% confluency.TEM
HMVECs were grown to 80% confluency in a 6-well plate. In the first experiment cells were incubated at 37 °C for 6 h with 1.6 μm and 3.2 μm hemispherical particles functionalized with either APTES or oxidation only. In the second experiment 3.2 μm discoidal particles were loaded with 15 nm SPIONs, amine-PEG coated, and subsequently incubated with the cells for 24 h. All samples were then washed with PBS and fixed with a solution of 2% parafomaldehyde (Electron Microscopy Science, Hatfield, PA) and 3% glutaraldehyde (Electron Microscopy Science, Hatfield, PA). After fixation, samples were washed and treated with 0.1% cacodylate-buffered tannic acid, and stained with 1% buffered osmium tetraoxide for 30 min followed by 1% uracyl acetate. Samples were then dehydrated in increasing concentration of ethanol and embedded in Poly-bed 812 medium. Samples were polymerized in a 60 °C oven for 2 days. Ultrathin sections were cut in a Leica Ultracut microtome (Leica, Deerfield, IL), stained with uracyl acetate and lead citrate in a Leica EM Stainer and examined in a JEM 1010 transmission electron microscope (JOEL, USA. Inc, Peabody, MA).Nocodazole
HMVECs were grown overnight to 80% confluence on 1.5 mm glass cover slips. Cells were incubated with 3.2 μm hemispherical silicon particles for 5 h in either regular medium or medium containing 150 nM nocodazole (Sigma). Cells were then washed with PBS, fixed in paraformaldehyde 4% for 10 min and permeabilized with 0.1% Triton X-100 for 3 min. PBS containing 1% BSA was used as a blocking agent prior to staining. Microtubules were stained using FITC-labeled monoclonal mouse anti-α-tubulin antibody (Abcam) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 dilution in PBS containing 1% BSA. Nuclei were stained using DRAQ5™ (Biostatus Ltd) at a 1:1000 dilution in PBS containing 1% BSA. Cells were washed with PBS and mounted on glass slides using Prolong Gold as the mounting solution. Images were taken using a Leica DM6000 upright confocal microscope equipped with a 63× immersion objective.
:200 dilution in PBS containing 1% BSA. Nuclei were stained using DRAQ5™ (Biostatus Ltd) at a 1:1000 dilution in PBS containing 1% BSA. Cells were washed with PBS and mounted on glass slides using Prolong Gold as the mounting solution. Images were taken using a Leica DM6000 upright confocal microscope equipped with a 63× immersion objective.
Flow cytometry
HMVECs were grown in a 6-well plate overnight to 80% confluence. 1.6 μm APTES-modified hemisherical particles were functionalized with a pH sensitive dye (pH Rhodo-NHS Ester, Invitrogen). The conjugation was performed by incubating the hemisherical particles in DMSO with the dye for two hours at room temperature, linking the carboxyl group on pH Rhodo to the amine group on the APTES modified particles. The particles were then washed two times with DMSO and two times with IPA. Cells were starved in serum-free medium for 40 min at 37 °C prior adding the pH Rhodo-modified silicon particles (1:10 cell-to-particle ratio). The plates were centrifuged for 4 min at 1500 rpm to allow the particles to settle down rapidly before incubating them at 37 °C for different amounts of time. Cells were detached using trypsin/EDTA solution and fixed with 4% parafomaldehyde. The samples were then analyzed with a Becton Dickinson FACS Calibur equipped with a 488-nm Argon laser and CellQuest software.
GFP-NPC1 transfection
Transfections were performed using an Amaxa Electroporator and the AMAXA HUVEC Nucleofactor kit (Lonza). Cells were removed with trypsin and counted. 1 × 106 HMVECs were aliquoted, centrifuged and resuspended in 100 μL of Nucleofactor solution, at room temperature. The 100 μL cell suspension was combined with 5 μg DNA (the GFP-NPC1 construct was a kind gift from Dr Junghae Suh at Rice University). The cell/DNA solution was transferred into certified a cuvette, paying attention not to trap air bubbles. The cuvette was inserted in the holder and the appropriate Nucleofactor program for HMVEC was applied. The cuvette was then taken out and 500 μL of a pre-warmed medium was added into it. The cells were immediately plated into a 24-well glass bottom dish and allowed to adhere overnight.Three different types of particles were employed in the experiment: 3.2 μm 594-dylight (Pierce) conjugated hemispherical, 3.2 μm and 1.6 μm APTES-modified hemispherical particles. Particles were incubated with the transfected cells for 2 h, then washed with phosphate buffer and fixed with paraformaldehyde 4%. Images were taken with a 1 × 81 Olympus Microscope with a 40× objective.
Live imaging
HMVECs were plated at 25![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 cells per well in 24-well plates with glass bottoms (MatTek Corporation, Ashland, MA) and allowed to adhere and grow overnight. Silicon particles were introduced after 24 h and the cells were visualized with a phase contrast microscope equipped with a humidified 37 °C incubator with 5% atmospheric CO2 (1 × 81 Olympus Microscope). A 20× objective was used and 5 photographs of different focal planes were taken at 5 min intervals for 19 h. Both 1.6 μm and 3.2 μm oxidized or APTES-modified hemispherical silicon particles were used. Spherical silica beads included three sizes, 1 μm, 2.5 μm and 3 μm. Three independent experiments were performed.
000 cells per well in 24-well plates with glass bottoms (MatTek Corporation, Ashland, MA) and allowed to adhere and grow overnight. Silicon particles were introduced after 24 h and the cells were visualized with a phase contrast microscope equipped with a humidified 37 °C incubator with 5% atmospheric CO2 (1 × 81 Olympus Microscope). A 20× objective was used and 5 photographs of different focal planes were taken at 5 min intervals for 19 h. Both 1.6 μm and 3.2 μm oxidized or APTES-modified hemispherical silicon particles were used. Spherical silica beads included three sizes, 1 μm, 2.5 μm and 3 μm. Three independent experiments were performed.
Particle tracking
Microparticle migration was analyzed by tracking their x and y movements using Slide Book software. The coordinates of the particles were transformed into time-averaged mean square displacement (MSD),| 〈Δr2(τ)〉 = 〈[x(t + τ) − x(t)]2 + [y(t + τ) − y(τ)]2〉 |
Quantification of nanoparticle exocytosis
HMVECs were grown overnight to 80% confluency on a 6-well plate in 1 mL of medium. The cells were then incubated at 37 °C with 3 × 106 loaded discoidal particles per well or with 5 μg of SPIONs. Supernatant was removed after 12 h and fresh medium was added to all samples. This time point was considered our time zero and the amount of iron present in the removed supernatant, not yet internalized, was subtracted from the calculated amount of iron added to cells. Medium and trypsinized cells were collected after 1, 3, 5 and 7 days of incubation. Cells were split after 3 days and measurements from both medium and cell pellets were recombined for the following analysis. Collected samples were centrifuged at 4200 rpm for 30 min and the supernatant was removed. Samples were dissolved by addition of 10 μL of 10–12 M hydrochloric acid then moved to a thermomixer set to 60 °C and 1300 RPM for 2 h. After dissolution, a 1% Spectrosol solution (CFA-C, Lot No. 111808, Spectrasol Inc.) brought to pH 8.5 with concentrated nitric acid was used to bring the sample volume to 5 mL, and 50μL of a 100 mg L−1 solution of yttrium was added to each sample and standard. Samples were analyzed for iron and silicon using axial ICP-OES with iron standard concentrations of 10, 25, 50, 100, 250, and 1000 mg L−1, silicon standard concentrations of 50, 100, 250 and 500 mg L−1, one blank sample of 1% Spectrosol solution, and one quality control standard containing 125 mg L−1 iron and 125 mg L−1 silicon. The instrument used for ICP-OES is a Varian Vista AX at a power of 1 kW, with plasma flow set to 15 L min−1, auxiliary flow of 1.5 L min−1 and a nebulizer flow of 0.75 L min−1, with 5 replicate readings at 15 s between each reading.Acknowledgements
The authors are grateful to Kenneth Dunner Jr. at MD Anderson Cancer Center (High Resolution Image Facility) for sample processing and analysis by transmission electron microscopy, to Jared K. Burks for confocal imaging performed at the Flow Cytometry and Image Facility (UT-MDACC) and Glen Snyder at Rice University for ICP analysis. A special thanks to our colleagues, Jeff Schmulen and Jean R. Fakhoury, at the University of Texas at Austin for silicon particle fabrication. This research was supported by the Department of Defense grants DODW81XWH-07-1-0596 and DODW81XWH-09-1-0212; NASA NNJ06HE06A; NIH RO1CA128797, U54CA143837, MH58920, and RC2GM092599; MDACC Institutional Core Grant #CA-016672; The State of Texas Emerging Technology Fund.References
- M. Ferrari, Trends Biotechnol., 2010, 28, 181–188 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS.
- E. Tasciotti, X. Liu, R. Bhavane, K. Plant, A. D. Leonard, B. K. Price, M. M.-C. Cheng, P. Decuzzi, J. M. Tour, F. Robertson and M. Ferrari, Nat. Nanotechnol., 2008, 3, 151–157 CrossRef CAS.
- P. Decuzzi and M. Ferrari, Biomaterials, 2008, 29, 377–384 CrossRef CAS.
- P. Decuzzi and M. Ferrari, Biomaterials, 31, 173–179 Search PubMed.
- R. M. Sawant, J. P. Hurley, S. Salmaso, A. Kale, E. Tolcheva, T. S. Levchenko and V. P. Torchilin, Bioconjugate Chem., 2006, 17, 943–949 CrossRef CAS.
- R. E. Serda, S. Ferrati, B. Godin, E. Tasciotti, X. Liu and M. Ferrari, Nanoscale, 2009, 1, 250–259 RSC.
- R. E. Serda, J. Gu, R. C. Bhavane, X. Liu, C. Chiappini, P. Decuzzi and M. Ferrari, Biomaterials, 2009, 30, 2440–2448 CrossRef CAS.
- R. E. Serda, J. Gu, J. K. Burks, K. Ferrari, C. Ferrari and M. Ferrari, Cytometry, Part A, 2009, 75a, 752–760 CrossRef CAS.
- S. Muro, M. Koval and V. Muzykantov, Curr. Vasc. Pharmacol., 2004, 2, 281–299 Search PubMed.
- M. J. Clague, Biochem. J., 1998, 336, 271–282 CAS.
- K. K. Huynh, E. L. Eskelinen, C. Scott, A. Malevanets, P. Saftig and S. Grinstein, EMBO J., 2007, 26, 313–324 CrossRef CAS.
- A. Blocker, F. F. Severin, A. Habermann, A. A. Hyman, G. Griffiths and J. K. Burkhardt, J. Biol. Chem., 1996, 271, 3803–3811 CrossRef CAS.
- A. Blocker, G. Griffiths, J. C. Olivo, A. A. Hyman and F. F. Severin, J. Cell Sci., 1998, 111, 303–312 CAS.
- R. E. Harrison, C. Bucci, O. V. Vieira, T. A. Schroer and S. Grinstein, Mol. Cell. Biol., 2003, 23, 6494–6506 CrossRef CAS.
- J. Suh, D. Wirtz and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3878–3882 CrossRef CAS.
- C. Wilhelm, F. Lavialle, C. Pechoux, I. Tatischeff and F. Gazeau, Small, 2008, 4, 577–582 CrossRef CAS.
- H. Duan and S. Nie, J. Am. Chem. Soc., 2007, 129, 3333–3338 CrossRef CAS.
- J. Panyam, W. Zhou, S. Prabha, S. Sahoo and V. Labhasetwar, FASEB J., 2002, 16, 1217–1226 CrossRef CAS.
- N. Fang, V. Chan, H. Mao and K. Leong, Biomacromolecules, 2001, 2, 1161–1168 CrossRef CAS.
- K. Bowman and K. W. Leong, Int. J. Nanomed., 2006, 1, 117–128 CrossRef CAS.
- J. Panyam and V. Labhasetwar, Pharm. Res., 2003, 20, 212–220 CrossRef CAS.
- C. Chiappini, E. Tasciotti, J. R. Fakhoury, D. Fine, L. Pullan, Y.-C. Wang, L. Fu, X. Liu and M. Ferrari, ChemPhysChem, 2010, 11, 1029–1035 CAS.
- R. Serda, A. Mack,A. van de Ven, S. Ferrati, B. Godin, C. Chiappini, K. Dunner Jr., M. Landry, L. Brousseau, X. Liu, A. J. Bean and M. Ferrari, submitted, 2010.
Footnotes |
| † Electronic supplementary information (ESI) available: Confocal microscopy image showing internalized negative particles, and movie of the intracellular migration of silicon particles. See DOI: 10.1039/c0nr00227e |
| ‡ Shared senior authorship |
| This journal is © The Royal Society of Chemistry 2010 |