Water soluble nanoporous nanoparticle for in vivo targeted drug delivery and controlled release in B cells tumor context†
F.
De Angelis‡
ab,
A.
Pujia‡
a,
C.
Falcone‡
c,
E.
Iaccino‡
c,
C.
Palmieri
c,
C.
Liberale
ab,
F.
Mecarini
a,
P.
Candeloro
b,
L.
Luberto
c,
A.
de Laurentiis
c,
G.
Das
a,
G.
Scala
c and
E.
Di Fabrizio
*ab
aFondazione Istituto Italiano di Tecnologia, Via Morego 30, 16163, Genova, Italy. E-mail: enzo.difabrizio@iit.it
bBIONEM Lab, University of Magna Graecia, viale Europa, 88100, Catanzaro, Italy
cDepartment of Experimental and Clinical Medicine, University of Magna Graecia, viale Europa, 88100, Catanzaro, Italy
First published on 7th September 2010
Abstract
Multitasking nanoparticles are gaining great attention for smart drug delivery systems. The exploration of the nano-scale opens new concrete opportunities for revealing new properties and undiscovered cell–particle interactions. Here we present a biodegradable nanoporous silicon nanoparticle that can be successfully employed for in vivo targeted drug delivery and sustained release. The bare nanoporous nanocarriers can be accurately designed and fabricated with an effective control of porosity, surface chemistry and particle size, up to a few nm. The proposed nanoparticles exhibit several remarkable features including high payload, biodegradability, no toxicity, and multiple loading in water without the need of additional chemical reagents at room temperature. The targeting strategy is based on phage display technology that was successfully used to discover cell surface binding peptide for murine B lymphoma A20 cell line. The peptide used in combination with the nanoporous nanoparticles allows an efficient in vivo targeting, a sustained release and a sensible therapeutic effect.
Introduction
Nanocarrier mediated drug delivery is one of the most attracting application of the emerging Nanomedicine. Targeted drug delivery and temporally controlled release improve the bioavailability of a drug reducing collateral effects and enhancing pharmacological and therapeutic properties. The basic idea is to deliver the drug only where it is needed, in proximity to or inside a cancer cell, and to release a desired quantity of drug over a controllable period of time. Several targeting strategies relying on antibodies, proteins, and peptides1–4 were studied, and their coupling with novel nanocarriers represents an open task for modern nanomedicine. To this end, different approaches are being developed for different nanomaterials, such as polymer–drug conjugates, liposomes, polymeric nanoparticles, micelles, and dendrimers, which represent the powerful arsenal of nanocarriers.5 Despite some differences, all drug delivery strategies based on nanocarriers rely on the same basic principle: drug and targeting agent are bound to the same vector that reaches the target and delivers the drug over a controlled period of time. Beside the discovery of novel and powerful drugs and targeting agents, there are still many difficulties to overcome in order to create an ideal nanocarrier. Some of the most important challenges are the conjugation with the host molecules (because they usually exhibit different chemical properties), multiple loading, biodegradability and toxicity.5–7 Moreover, 40% of anticancer drug candidates suffer from poor solubility,7 which can be overcome in a nanoporous carrier where the limited space of the pores prevents the formation of the crystal phase, resulting in a higher solubility of drug. Among a lot of intense studies, porous silicon films and micro-particles have been proposed for biomedical applications,8–12 showing several remarkable features such as high loading and releasing capacity, protection from body's natural immunological responses, in vivo monitoring, easy and well known chemistry, and low toxicity. Morphology and pore size distribution can be finely controlled, together with the surface chemistry, to allow a flexible control of bioreactivity and hydrophobicity.12–14 These promising studies were carried out on micrometre sized particles, but it's possible to speculate that nanoparticles with diameters between 20–80 nm would be optimal for cancer therapy.6,15–17 In fact, particles in this range are small enough to penetrate tumor vessel pores, large enough to avoid renal filtration, but not too large to induce transient poration of the cell membrane and cytotoxicity.In the near future a lot of different nanocarriers are expected to be proposed, and this complex scenario will force the scientific community toward a laborious case-by-case evaluation. Then, it would be desirable to introduce a polyvalent, biodegradable, and inert carrier, able to easily embed many molecules without interacting with them, to treat a wide range of diseases both under a diagnostic and therapeutic point of view. To this end, all of these nano-abilities will be exploited in the next future to rise to the challenge of developing a nano-craft that will have the potential to revolutionize cancer treatments, improving drug adsorption, distribution, and release profiles.
Here we present a water soluble nanoporous silicon nanoparticle able to fulfil all of the most important requirements of an ideal nanocarrier, and then they can be candidate as one of the simplest building block for the forthcoming nanocrafts.
In the following, we first show the basic features of the bare nanoparticles, and second, we introduce a tumour-specific peptide able to perform both an efficient tumor-targeting and a therapeutic effect.18 The reason for peptide introduction is twofold: on one hand it represents a valid therapeutic approach; on the other hand, it demonstrates that the nanocarrier is suitable for practical applications. In fact, when used in combination with peptides, the nanoporous nanoparticles allow an efficient in vivo targeted drug delivery and controlled release. A set of experiments concerning release profile, biodistribution, toxicity, imaging ability, and therapeutic effects, are presented both in vitro and in vivo.
Results
Nanoporous nanoparticles fabrication and characterization
Nanoporous silicon nanoparticles (NPNPs) were fabricated by ultra-sonication of a thin film of porous silicon: the fabrication process takes a few steps which can be easily scaled for industrial production (the whole process, including drug loading, is pictorially described in Fig. 1, for further details see ESI section 1†). Particle sizes can be adjusted in the range from 5 nm up to 500 nm, varying time and power of ultra-sonication process (ESI section 1†). Electron Scanning Microscope images of NPNPs with a diameters of about 50 and 200 nm are shown in Fig. 2 (panel A and B). SEM image with a magnification of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 (panel C), shows the porous structure of silicon sample (cross-section). The silicon nanocrystals that constitute the aggregate, have a diameter of about 5–7 nm, and the nanocrystals inter-spaces are related to the degree of porosity of material that is available to load host molecules. The pore size of NPNPs is too small to be well evidenced with the electron scanning microscope (SEM), but the characteristic luminescence band, with an emission spectrum centred at around 675 nm (UV illumination, panel D) suggests a pore size of about 2 nm (compatible with SEM observation). When placed in physiological solution, the NPNPs dissolve11 through a progressive disjointing of the silicon nanocrystal skeleton, resulting in a release of the host molecules. The disjointed silicon nanocrystals (5 nm diameter) are non-toxic10 and they are rapidly cleaned by renal filtration.
000000 (panel C), shows the porous structure of silicon sample (cross-section). The silicon nanocrystals that constitute the aggregate, have a diameter of about 5–7 nm, and the nanocrystals inter-spaces are related to the degree of porosity of material that is available to load host molecules. The pore size of NPNPs is too small to be well evidenced with the electron scanning microscope (SEM), but the characteristic luminescence band, with an emission spectrum centred at around 675 nm (UV illumination, panel D) suggests a pore size of about 2 nm (compatible with SEM observation). When placed in physiological solution, the NPNPs dissolve11 through a progressive disjointing of the silicon nanocrystal skeleton, resulting in a release of the host molecules. The disjointed silicon nanocrystals (5 nm diameter) are non-toxic10 and they are rapidly cleaned by renal filtration.
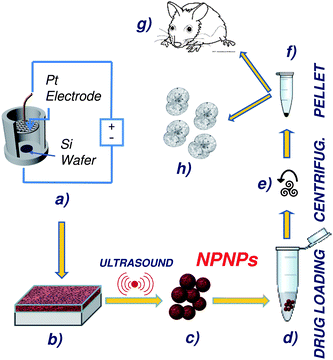 | ||
| Fig. 1 Schematic representation of the overall process, from nanoparticle fabrication, to drug loading and administration. (a, b) Nanoporous silicon film is obtained from a silicon wafer through a well-defined electrolytic process; thanks to a controllable employing of ultrasound, the nanoporous film can be disjointed to obtain nanoporous nanoparticles of desired size (c). After filtration, host molecules are loaded on the nanoparticles by incubation (d), and the excess of drug can be recovered by centrifugation (e). The obtained nanoparticles with drug embedded (pellet, (f)) can be stored or administrated (g, h). | ||
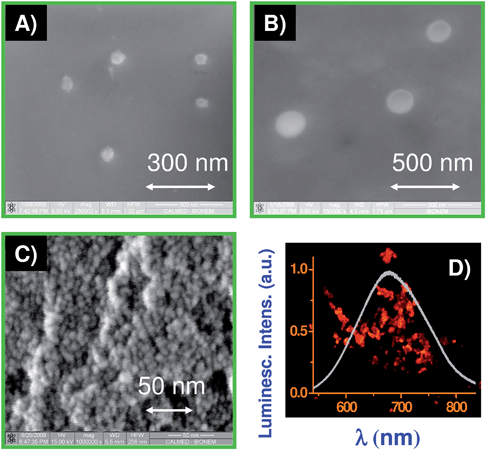 | ||
| Fig. 2 Nanoporous silicon nanoparticles: SEM images of NPNPs of 50 and 200 nm diameter (A, B); (C) SEM image (×106 magnification) of porous silicon cross section showing the skeleton structure made of silicon nanocrystals with a diameter of 5–7 nm. The interspaces between nanocrystal define the free volume available to load host molecules. (D) Optical image of the NPNP and related emission spectrum, when they are illuminated with UV light. | ||
Multivalent drug loading and release profiles
Concerning drug delivery, our approach consists of loading the drug into the nanopores and controlling the drug release over a period of a few days. As stated above, the most interesting range of size for cancer therapy is 20–80 nm then we mainly focused our attention on 50 nm NPNPs.In order to evaluate the ability of NPNPs to adsorb small molecules they were loaded with different low molecular weight species, and analysed by fluorimetry. The loading process involves two steps: a short incubation (1 h); followed by a centrifugation (ESI section 2–4†). During the incubation, small molecules (below 15 kDa in weight) can diffuse inside the pores where they are trapped by physi-sorption.10,19 Since no covalent bonding occurs, the chemical properties of the payload remain unaltered.
The whole process can be carried out in physiological solution at room temperature, without the need to introducing particular chemical reactants, which can degrade the drug, or contaminate the nanocarrier, induce toxicity, or other unwanted properties that require a more complicated clinical study. Moreover, drug loading at room temperature enables the use of NPNPs also with sensitive therapeutic compounds susceptible of degradation, like some peptides and proteins.14 The excess of drug (fraction not adsorbed on the NPNPs during incubation), can be easily recovered by means of centrifugation resulting in a reduction of production waste, that is very important in case of expensive or toxic drugs.
In Fig. 3 (panels A and B), a set of Confocal Microscopy images of NPNPs with two fluorophores simultaneously embedded, are reported as a proof of bivalent loading (Fluorescein disolved in NaCOH3 and Alexa Fluor® 405 cadaverine dissolved in H2O were mixed in ratio fluorescein/cadaverine = 1/3, and then incubated with the NPNPs). Indeed, more than 20 molecular species were simultaneously loaded, without significantly affecting the dissolution rate of the NPNPs (ESI section 4†). In addition to the remarkable capability of multivalent loading, the NPNPs exhibit an extremely high loading capacity of about 3 × 10−17 g of drug per each 50 nm NPNP (this value is about 50% of maximum loading capacity of a 50 nm nanosphere, see also ESI section 3†). The release profile of Fluorescein loaded on NPNPs (50 nm) is shown in Fig. 3C: under physiological conditions (PBS solution, 37 °C) the NPNPs dissolve releasing the payload in about 120 h. In case of administration, a fraction of the payload is released in the blood stream before encountering the target, but being the dissolution time so long (several hours) this fraction is irrelevant. However, being dependent on the pore size and environmental conditions, the dissolution rate can be accurately adjusted, and can take from a few hours to a few weeks.
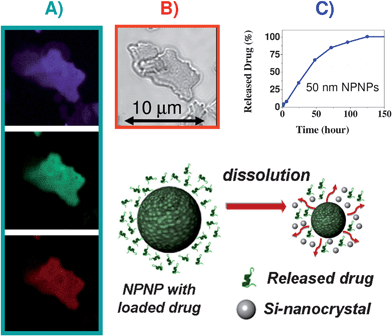 | ||
| Fig. 3 Multivalent drug loading and release of the NPNPs. (A, B) Confocal images of a NPNPs with two fluorophores simultaneously embedded: in panel A, fluorescein signal, cadaverine signal, and intrinsic luminescence of the NPNP are visible on purple, green, and red channel, respectively. In panel B, the same sample of NPNPs dropped on a slide is shown on transmission channel. (C) Release profile under physiological condition (PBS solution, 37 °C) of fluorescein loaded on NPNPs. | ||
For pH values below 5, the NPNPs don't dissolve allowing long term storage (also with drug embedded) and transport without particular precautions (ESI section 3†).
In vitro functional outcome
As mentioned above and further discussed in the next section, we employed a tumor-specific peptide able to perform both targeted delivery and therapeutic effect, which can be enhanced through the combination with the NPNPs. The targeting strategy is based on phage display technology that, in the last decade, was successfully used to discover cell surface binding peptides.3,20–22 We identified an idiotypic-specific peptide, termed pA20-6, which binds with high affinity and specificity the sIg of A20 cells both in vitro and in vivo; moreover, pA20-6 could be internalized and induce a therapeutic effect due to cell cycle arrest and apoptosis.18 Hence, pA20-6 peptide was selected to investigate the nanoporous nanoparticles potentiality.To evaluate in vitro functional outcome, NPNPs and peptide (FITC-pA20-6) were incubated with cultured A20 cells and monitored by flow cytometry over a period of 72 h. Five different cases were studied: pA20-6 alone, NPNPs of 50 with and without peptide, NPNPs of 200 nm with and without peptide. The measured uptakes on A20 cells are reported in Fig. 4A. The results confirm that the pA20-6 alone is able to reach the tumor cell, although its binding ability is limited and it slowly decreases after 24 h. As can be clearly seen, when drug delivery is mediated through the NPNPs, a more efficient targeting and uptake occur over the first 24 h, and during this time the uptake increased sevenfold. After 24 h the targeting/uptake profiles of the peptide with and without the help of NPNPs are very similar, probably because in both cases the uptake of the peptide is dominated by surface receptor turn-over mechanism (the disappearing of surface bound peptide as Ig-BCR engagement results in antigen uptake by BCR-mediated endocytosis). We also notice that, as expected, the smaller NPNPs (50 nm) exhibit a more efficient uptake compared with the bigger one (200 nm).
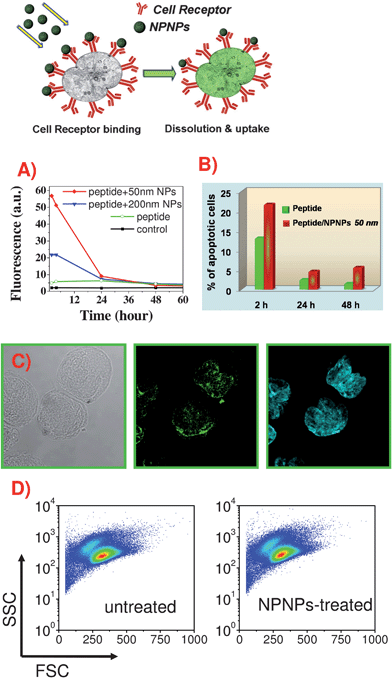 | ||
| Fig. 4 In vitro targeting ability, therapeutic effects, and toxicity of the peptide/NPNPs. (A) Flow cytometry analysis of uptake on A20 cells of pA20-6-FITC with and without the help of NPNPs of different sizes. (B) Apoptotic effect of pA20-6 on A20 cells with and without the help of nanocarrier (calculated through cell cycle analysis). (C) Confocal images showing the binding of pA20-6-FITC/NPNPs on A20 cells. From left to right: transmission, green, and cyan channels show A20 cells, pA20-6-FITC/NPNPs distribution, and nuclear staining (Hoechst S769121, Invitrogen), respectively. (D) FSC/SSC (front/side scattering) plot of A20 cells treated with pA20-6/NPNPs and untreated. Between the two samples there aren't any differences, indicating that NPNPs did not affect cell morphology and viability (no apparent toxicity). | ||
Cell cycle analysis indicated an increased number of cells with a sub-G1 DNA (apoptotic) upon treatment with pA20-6, according to previous data,23,24 demonstrating that idiotype-specific peptides induced a therapeutic effect due to cell cycle arrest and apoptosis of the tumor cell. In Fig. 4B, the percentage of the apoptotic cells, obtained in vitro for free peptide and NPNPs mediated delivery are reported. As expected, when nanocarriers are used the bioavailability of peptide increases at tumor site (Fig. 4A) resulting in a more consistent apoptotic effect. The advantage of the NPNPs mediation is still evident in the period 24–48 h, even if the availability of the peptide, with and without NPNP, is comparable (Fig. 4B). In fact, in this range, a more evident apoptotic effect occurs when NPNP are exploited (Fig. 4C), probably thanks to the larger uptake occurred in the first 24 h.
In order to further verify the ability of peptide/NPNPs to reach selectively the target cancer cell, the complex was incubated with both A20 and Hela cells (epithelial cervix carcinoma). Confocal Microscopy analysis indicated that the NPNPs tend to localize only at the A20 cell surface, increasing the availability of peptide in close proximity of cells, and resulting in a higher binding ability and apoptosis (Fig. 4C). The proposed nanocarrier specifically recognizes the target cancer cells, whereas HeLa cells seem not interacting with the peptide (ESI section 6†).
The toxicity of the NPNPs was evaluated in vitro in different ways, and all experiments indicated no apparent toxicity: (1) NPNPs alone do not affect cell morphology and viability, as indicated by unchanged forward and lateral light scattering profiles of cells over time, reported in Fig. 4D (FSC/SSC plot after 24 and 48 h of incubation); (2) no evidence of secretion of the inflammatory cytokine IL1-β was found (ESI section 7†).
In vivo targeting, biodistribution, therapeutic effect, and toxicity
Finally, we evaluated in vivo targeting, biodistribution, and toxicity of the proposed nanocarrier, exploiting an A20 tumor model in syngenic mice. To this end, A20 cells were engrafted Balb/c in mice and, when they developed a palpable tumor mass, they were injected with pA20-6-FITC/NPNPs complex or with free peptide alone. After 1 and 4 h, mice were sacrificed and tumor, liver, kidney, and spleen were removed for further investigation. Histological analyses of tumor tissues are shown in Fig. 5, and the results confirm those obtained in vitro: it is evident that pA20-6 is able to target the cancer cells but the binding tends to vanish within a few hours. On the contrary, the use of the NPNPs allow in vivo sustained binding and release without compromising the efficiency of the targeting. To quantify the enhancement due to the NPNPs employment, the biodistribution of the pA20-6, with and without the help of the nanocarriers, was investigated by flow cytometry analysis after 4 h from the administration (Fig. 6). The percentages of cells positive for peptide binding are indicated in the graphs: in the case of free peptide administration, the percentage of tumor cells reached by the drug is about 8% (3% for metastatic cells in spleen). When the drug delivery is mediated through the nanocarrier, the bioavailability of the peptide increases, and the efficacy of the targeting rises up to 3-fold with respect to free peptide administration, without affecting other tissues (liver and kidney).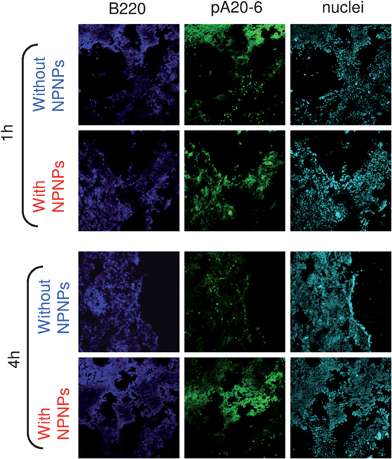 | ||
| Fig. 5 In vivo targeting. Histological analyses of tumor tissues in Balb/c mice treated with pA20-6 with and without the help of nanocarriers. Upper panels: 1 h after peptide injection. Bottom panels: 4 h after peptide injection. Blue channel: B220 FITC antibody revealing tumor cells. Green channel: FITC-pA20-6/NPNPs. Cyan channel: nuclei staining. | ||
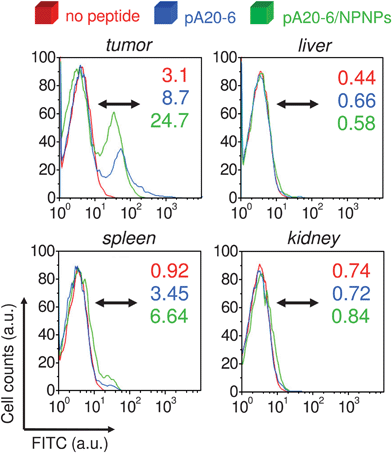 | ||
| Fig. 6 In vivo biodistribution. Flow cytometry analyses of different tissues of Balb/c mice treated with pA20-6 with and without the help of nanocarriers. The percentages of cells, positive for peptide binding, are indicated in graphs (after 4 h from the administration). | ||
As mentioned above, in vivo toxicity were assessed, by evaluating serum levels of both inflammatory cytokine IL1-β and hepatotoxicity markers LDH and GSH. No abnormal values of these markers were observed in BALB/c mice after repeated NPNPs administrations (ESI section 8†), or obvious histological evidences of tissue pathology in liver, kidney, spleen, lungs and heart. Moreover, mice treated with NPNPs did not shown any sign of systemic toxicity as estimated by body weight and gross appearance.
Discussion
Peptides are powerful molecular tools for cell targeting due to their small size and functional versatility, including multivalency for the targeting moiety and drug attachment. Peptides that are N- and C-terminally blocked may acquire a cyclic, constrained conformation, and may include unnatural and D-amino acids with extended half-life in vivo.25,26 Examples of tumor-specific peptides with diagnostic and therapeutic potential include naturally occurring peptides, such as somatostatin and bombesin/gastrin-releasing peptide (BN/GRP), which exhibit extremely high affinities for cell-surface receptors. It has been also demonstrated that peptide facilitate the transport of cytotoxic compounds and nucleic acid into specific tumor tissues.3 Based on this evidence, we constructed and screened a random peptide library (RPL) displayed on f88-bacteriophage,27,28 in order to select peptides for the idiotypic determinants of surface immunoglobulins (sIg) of the murine B lymphoma A20.The vast majority of B lymphoma cells express antigen receptors composed of surface immunoglobulins within the B cell receptor (BCR). Since the B-cell tumors are clonal offspring of a single transformed lymphoid progenitor cell, with few exceptions the antigen receptors of lymphoma cells in an individual patient are identical and each receptor has a unique antigen-binding site. This unique immunoglobulin in lymphoma cells is commonly defined as ‘idiotype’, and it has been proposed as a potential target for immunotherapy over 25 years ago.29
Then we selected the pA20-6 peptide and we used it in combination with the NPNPs in order to show that our nanocarrier is actually suitable for practical application. The overall approach is easy and powerful: the tumor specific peptide plays the double role of targeting molecule and therapeutic agent, and the employment of the nanoporous nanorpaticles allows an enhancement of peptide features together with a sustained release.
We described an easy, cheap, and fast fabrication process with an accurate control of pore size and morphology of the nanocarrier. The most interesting particle size range for cancer therapy (20–80 nm) is fully accessible, accompanied by the well defined and powerful chemistry of silicon that make available hundreds of cross-linkers. Under physiological condition the NPNPs dissolve and release the loaded molecules over a period of a few days. The dissolution products are mainly composed of non toxic silicon nanocrystals9 which are cleaned by renal filtration.
Since NPNPs don't dissolve when suspended in aqueous acid ambient (pH < 5), no leaching of loaded molecules occurs under this condition. Thus, the pH value can act as a trigger to start or to stop the release: this is an important feature that enables stable long term storage before administration.
Through a spontaneous physisorption, a wide range of drugs can be loaded in water, at room temperature, without covalent bonding, and additional chemical reactants. Moreover, the excess of molecules not adsorbed during the incubation can be recovered by means of centrifugation.
Thanks to the capability of multivalent loading and high loading volume, the NPNPs can embed different molecules for multi-tasking applications (targeting, imaging, delivery), without cross talk between different tasks. In fact, the NPNPs can be also employed for imaging experiments, as demonstrated by confocal analyses reported in Fig. 4C. Modern medicine asks not only for fast and easy techniques but strongly demands for a fully compatibility with existing protocols. Thanks to its versatility, the proposed nanocarrier can be mixed and matched with the majority of current targeting and therapeutic protocols.
Through a set of experiments performed both in vitro and in vivo we demonstrated that the NPNPs increase the bioavailability of the peptide and then enhances both targeting and therapeutic effects. Finally, no apparent toxicity was evidenced both in vitro and in vivo.
The presented results together with the data already reported in literature, candidates these nanoparticles as building-block of the forthcoming generation of nanocrafts.
Materials and methods
NPNPs fabrication
Porous silicon was obtained by anodization of boron-doped silicon wafer (resistivity 5–10 Ω cm) of [100] crystal orientation, using an electrolyte mixture of hydrofluoric acid (25%), water (25%), and ethanol (50%). Applied constant current density: 10 mA cm−2 for 5 min at 25 °C. Samples were rinsed in deionized water, then in ethanol, and pentane. The nanoporous film was dried in oven at 260 °C for 4 h. In order to obtain NPNPs, the porous silicon film were sonicated in DMF (Dimethylformamide) for 60 min and then, after washes in ethanol, ultra-sonicated (5 W) in water for 10 min at a constant temperature of 4 °C, and finally filtered to eliminate impurities above 500 nm.Affinity selection using random peptide phage libraries
To select epitopes specifically recognized by surface immunogobulins from tumor B cells, a 15-mer phage-display random peptide library (f88-4/Cys6, 1.87 × 1011 peptide sequences displayed on the amino-terminus of the pIII protein of the filamentous bacteriophage fd-tet) was screened with the monoclonal antibody (mAb-A20) isolated from culture of the murine B lymphoma A20 as described.30 In the immunoaffinity selection, 20 μg of mAb A20 was immobilized to streptavidin-conjugated magnetic beads (Promega) previously coated with 10 μg of biotinylated anti-mouse IgG F(ab)2-specific polyclonal Ab (Sigma) in 200 μl of beads suspension. A total of 1 × 1011 of transducing units of bacteriophages from the libraries were added and incubated for 12–16 h at 4 °C. After extensive washing, bound phages were eluted with 0.1 M HCl/glycine buffer (pH 2.2) and neutralized with 1M Tris pH 9.1 and amplified in ER2738 cells for further rounds of panning. ELISA binding assay on purified phages was performed to select clones positive for the mAb A20. Peptide synthesis was obtained from CASLO Laboratory ApS, Technical University of Denmark.In vivo pA20-6/NPNPs tumor targeting and bio-distribution
Female BALB/c mice were obtained from Harlan (Indianapolis, IN, USA) at 6 to 8 weeks.3 groups of Balb-C mice were engrafted sub-cutis with 2 × 106 A20 cells and, when tumor masses became palpable, mice were i.v. injected with pA20-6-loaded NPNPs (50 μg of peptide loaded on 1.6 × 1012 nanoparticles), with free peptide (50 μg) or with PBS (control mice group). After 4 h, mice were sacrificed and tumor, liver, spleen and kidneys were removed. Single cells suspension from these organs were prepared and investigated by flow cytometry for peptide binding.
Concerning flow cytometry of Fig. 6: the frozen tissue was thawed on ice. A cell strainer (FALCON) was placed on top of a 50-ml Falcon tube, the tissue was placed into the strainer, and 0.5 ml of PBS was added. Gentle pressure was applied, and the tissue was squeezed through the cell strainer, which resulted in a single-cell suspension. More PBS was added to rinse the cell strainer up to a total volume of 5 ml. Depending on the cell density, 50–100 μl of this suspension were used for analysis.
Fresh sections from tumor masses were prepared and subjected to histological analysis. In particular, tumor sections were stained with B220 FITC antibody (BD Pharmingen) and fixed in Cytofix/Cytoperm Solution (BD Pharmingen) for 30′ at 4 °C; nuclei staining was performed with DAPI and slides were examined by confocal microscopy (Leica, TC-SP2).
Acknowledgements
We thank Dr Lisa Vaccari and Dr Marco Gaspari for their helpful suggestions. This work was funded under European Project DIPNA FP6-STREP proposal no. 032131, and Project SMD FP7-NMP 2800-SMALL-2 proposal no. CP-FP 229375-2.References
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Active targeting schemes for nanoparticle systems in cancer therapeutics, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS.
- R. Weissleder, K. Kimberly, E. Yi Sun, T. Shtatland and L. Josephson, Cell-specific targeting of nanoparticles by multivalent attachment of small molecules, Nat. Biotechnol., 2005, 23, 1418–1423 CrossRef CAS.
- S. M. Okarvi, Peptide-based radiopharmaceuticals and cytotoxic conjugates: potential tools against cancer, Cancer Treat. Rev., 2008, 34, 13–26 CrossRef CAS.
- C. Palmieri, et al., In vivo targeting and growth inhibition of the A20 murine B-cell lymphoma by an idiotype-specific peptide binder, Blood, 2010, 116, 226, DOI:10.1182/blood-2009-11-253617.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nanocarrier as an emerging platform for cancer therapy, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- J. R. Heath and M. E. Davis, Nanotechnology and cancer, Annu. Rev. Med., 2008, 59, 251–265 CrossRef CAS.
- D. Putnam, Drug delivery, The heart of the matter, Nat. Mater., 2008, 7, 836–837 CrossRef CAS.
- J. Salonen, et al. Mesoporous silicon microparticles for oral drug delivery: Loading and release of five model drugs, J. Controlled Release, 2005, 108, 362–374 CrossRef CAS.
- J. Salonen, A. M. Kaukonen, J. Hirvonen and V. P. Lehto, Mesoporous silicon in drug delivery applications, J. Pharm. Sci., 2008, 97, 632–653 CrossRef CAS.
- E. J. Anglin, L. Cheng, W. R. Freeman and M. J. Sailor, Porous silicon in drug delivery devices and materials, Adv. Drug Delivery Rev., 2008, 60, 1266–1277 CrossRef CAS.
- L. Vaccari, et al. Porous silicon as drug carrier for controlled delivery of doxorubicin anticancer agent, Microelectron. Eng., 2006, 83, 1598–1601 CrossRef.
- Ji-Ho Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Biodegradable luminescent silicon nanoparticles for in vivo application, Nat. Mater., 2009, 8, 331–336 CrossRef CAS.
- A. M. Tinsley-Bown, et al. Tuning the pore size and surface chemistry of porous silicon for immunoassays, Phys. Status Solidi A, 2000, 182, 547 CrossRef CAS.
- T. Limnell, J. Riikonen, J. Salonen, A. M. Kaukonen, L. Laitinen, J. Hirvonen and V. P. Letho, Surface chemistry and pore size affect carrier properties of mesoporous silicon microparticles, Int. J. Pharm., 2007, 343, 141–147 CrossRef CAS.
- A. Verma, O. Uzun, Yuhua Hu, Ying Hu, Hee-Sun Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Surface-structure-regulated-cell-membrane penetration by monolayer-protected nanoparticles, Nat. Mater., 2008, 7, 588–595 CrossRef CAS.
- D. F. Emerich and C. G. Thanos, The pinpoint promise of nanoparticle-based drug delivery and molecular diagnosis, Biomol. Eng., 2006, 23, 171–184 CrossRef CAS.
- J. Lovrić, H. S. Bazzi, Yan Cuie, G. R. A. Fortin, F. M. Winnik and D. Maysinger, Differences in subcellular distribution and toxicity of green and red emitting CdTe quantum dots, J. Mol. Med., 2005, 83, 377–385 CrossRef.
- A. L. De Cerio, N. Zabalegui, M. Rodriguez-Calvillo, S. Inoges and M. Bendandi, Anti-idiotype antibodies in cancer treatment, Oncogene, 2007, 26, 3594–3602 CrossRef.
- S. H. C. Anderson, H. Elliott, D. J. Wallis, L. T. Canham and J. J. Powell, Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions, Phys. Status Solidi A, 2003, 197, 331–335 CrossRef CAS.
- R. Cortese, et al. Identification of biologically active peptides using random libraries displayed on phage, Curr. Opin. Biotechnol., 1995, 6, 73–80 CrossRef CAS.
- M. Graner, A. Raymond, E. Akporiaye and E. Katsanis, Tumor-derived multiple chaperone enrichment by free-solution isoelectric focusing yields potent antitumor vaccines, Cancer Immunol. Immunother., 2000, 49, 476–484 CrossRef CAS.
- K. J. Kim, C. Kanellopoulos-Langevin, R. M. Merwin, D. H. Sachs and R. Asofsky, Establishment and characterization of BALB/c lymphoma lines with B cell properties, J Immunol, 1979, 122, 549–554 CAS.
- M. F. Renschler, R. R. Bhatt, W. J. Dower and R. Levy, Synthetic peptide ligands of the antigen binding receptor induce programmed cell death in a human B-cell lymphoma, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3623–3627 CrossRef CAS.
- M. F. Renschler, H. G. Wada, K. S. Fok and R. Levy, B-Lymphoma cells are activated by peptide ligands of the antigen binding receptor or by anti-idiotypic antibody to induce extracellular acidification, Cancer Res, 1995, 55, 5642–5647 CAS.
- O. H. Aina, T. C. Sroka, M. L. Chen and K. S. Lam, Therapeutic cancer targeting peptides, Biopolymers, 2002, 66, 184–199 CrossRef CAS.
- O. H. Aina, R. Liu, J. L. Sutcliffe, J. Marik, Chong-Xian Pan and K. S. Lam, From combinatorial chemistry to cancer-targeting peptides, Mol. Pharmaceutics, 2007, 4, 631–651 CrossRef CAS.
- J. K. Scott and G. P. Smith, Searching for peptide ligands with an epitope library, Science, 1990, 249, 386–390 CrossRef CAS.
- G. Zhong, G. P. Smith, J. Berry and R. C. Brunham, Conformational mimicry of a chlamydial neutralization epitope on filamentous phage, J Biol Chem, 1994, 269, 24183–24188 CAS.
- G. T. Stevenson, E. V. Elliott and F. K. Stevenson, Idiotypic determinants on the surface immunoglobulin of neoplastic lymphocytes: a therapeutic target, Fed Proc, 1977, 36(9), 2268–71 Search PubMed.
- R. Cortese, F. Felici, G. Galfre, A. Luzzago, P. Monaci and A. Nicosia, Epitope discovery using peptide libraries displayed on phage, Trends Biotechnol., 1994, 12, 262–267 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Nanoparticles fabrication; payload evaluation; dissolution and release profiles; multivalent loading; targeting specifity on A20 Cells; cell cycle analysis; in vitro cytotoxicity assay; in vivo cytotoxicity assay. See DOI: 10.1039/c0nr00161a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2010 |