Structuration of pH-responsive fluorescent molecules on surfaces by soft lithographic techniques†
Alberto
Martínez-Otero
a,
Félix
Busqué
b,
Jordi
Hernando
*b and
Daniel
Ruiz-Molina
*a
aCentro de Investigación en Nanociencia y Nanotecnología (CIN2, CSIC-ICN), Esfera UAB, 08193, Cerdanyola del Vallès, Spain. E-mail: druiz@cin2.es; Fax: +34935813717; Tel: +34935814777
bDepartament de Química, Universitat Autònoma de Barcelona, 08193, Cerdanyola del Vallès, Spain. E-mail: jordi.hernando@uab.es; Fax: +34935811265; Tel: +34935814924
First published on 14th July 2010
Abstract
Two different soft lithographic techniques (LCW and μCP) have been successfully used for the structuration of fluorescent pH-responsive molecules on surface. The molecules of choice, fluorescein (1) and a new catechol derivative (2), exhibit several protonation states with distinct emission properties over a large acid–base range. This allowed us to fabricate fluorescent arrays that respond over a large pH-window.
Introduction
Nanostructuration of optical sensing materials on solid supports is being widely explored. When downscaling, the surface-to-volume ratio increases and surface properties become increasingly important, while requiring much less analyte, production and detection time.1 Moreover, both organization and miniaturization of sensing molecules on surfaces will allow the creation of denser and more complex combinatorial libraries. Such advantages have already led to significant advances in the development of sensor systems with promising applications in the detection of small biomolecules,2 metal ions,3 pH changes,4 organic vapors and explosives,5 as well as in medical diagnosis.6Among the different sensing capabilities that can be developed, we have focused our attention on the structuration of pH-responsive fluorescent molecules on surfaces. Previous works have already been reported on this topic with examples of pH-responsive molecules deposited on surfaces,4 optical fibers7 and even on cantilever arrays.8 However, in all cases sensing over large pH-windows still remains a challenge.
Two main approaches can be followed for the fabrication of large pH-window responsive fluorescent arrays. The first one relies on the use of two or more families of molecules whose emission properties respond over different pH ranges.9 Though effective, the combinatorial writing of more than one compound on the nanometre scale to prepare a miniaturized array of chemosensors still represents a real experimental challenge.10 The second approach, the one followed in this work, consists on the structuration of only one molecular system that itself exhibits multiple groups with different pKa values. Therefore protonation/deprotonation of such groups leads to multiple states with differential emission properties that respond over a large pH-window.4e, 11 According to the variation of their fluorescence response with pH, these systems can be classified into pH-induced ‘off–on–off’11e, 12 or ‘on–off–on’13 fluorescence switches.
One of such compounds is fluorescein (1), the most widely used extrinsic fluorescence probe in biosciences.14 In aqueous solution it occurs in cationic (c1), neutral (n1), anionic (a1) and dianionic (d1) forms (pKl = 2.08, pK2 = 4.31, and pK3 = 6.43, respectively), making its absorption and fluorescence properties strongly pH dependent (see Scheme 1). The dianion has the most intense fluorescence with a quantum yield of 0.93, whereas the fluorescence quantum yield of the anion is 0.37. On the contrary, the neutral and cationic species have fluorescence quantum yields close to 0.15 Using fluorescein, we have already fabricated ordered arrays down to micro and nanometre scales by using parallel Dip-Pen Nanolithography (DPN).16 Interestingly such arrays exhibited a pH-induced switching-like behavior related to that found in solution. Compound 1 interconverted between the neutral, anionic and dianionic forms exhibiting different emission properties in response to pH changes. This was one of the scarce examples that demonstrated the viability of multistate pH-fluorescent molecules on surfaces.
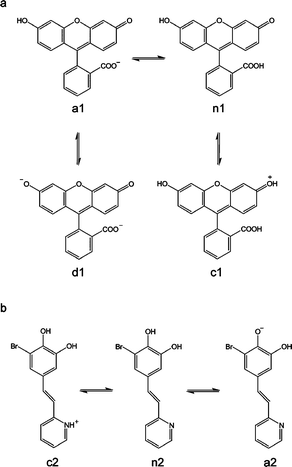 | ||
| Scheme 1 Acid–base equilibria of compounds 1 (a) and 2 (b). | ||
Inspired by this successful approach, in this work we have expanded the development of structured fluorescent pH-responsive molecular arrays on surfaces in a four-fold manner.17 First, we have deeply investigated their self-assembly mechanisms as well as surface-induced spectral shifts, both of their absorption and emission bands. Second, alternative soft lithographic techniques, Lithographically Controlled Wetting (LCW)18 and Micro-Contact Printing (μCP),19 have been used for the fabrication of the arrays on surfaces. In both cases soft stamps are used as one of the most versatile and technologically viable tools for micro- and nano-manufacturing over large scale areas. Third, structuration is achieved not only by direct deposition but also by grafting through covalent bonds. This will allow the use of the fabricated arrays for pH-sensing not only in gases but also under liquid environments, thanks to the considerable stability improvement. And fourth, we have added a new catechol derivative (2) that also acts as a multistate switchable molecule, for comparison purposes.20,21 Compound 2 has two groups presenting different acid–base activity, pyridine (pKa = 5.2 for the pyridinium ion in water) and 6-bromocatechol (pKa = 9.2 and 13.0 for pure catechol in water) (see Scheme 1), which gives rise to three different protonation states for this molecule: the cationic (c2), neutral (n2), and anionic (a2) forms, which can interconvert reversibly (see Scheme 1). The anionic form presents a fluorescence quantum yield of 0.22 and a maximum emission of fluorescence at 525 nm. The other two forms have fluorescence quantum yields under 0.07 and emission maxima at 511 nm (c2) and 409 nm (n2).21
Results and discussion
Optical properties
Some compounds show spectral shifts of their absorption and/or emission bands with respect to bulk solutions upon surface attachment or deposition.22 To investigate this issue, compounds 1 and 2 were deposited by spin-coating on a glass slide and examined by AFM scratching and fluorescence confocal microscopy. AFM scratching experiments allow the removal of materials of several nanometres in depth by AFM-scanning under strong loading forces.23 In our case, these experiments were used to analyze the layer thickness of the deposited compounds and its influence on the emission properties. As can be seen in Fig. 1, topographic AFM measurements done immediately after scratching over different 30 × 30 μm2 areas revealed a height profile for spin-coated layers of 1 of around 90 nm. Simultaneously, the emission properties of such a fluorescein thin-film were also studied by confocal fluorescence microscopy. Fig. 2a plots the fluorescence spectrum registered for the dianion form of 1 on glass, together with that obtained for an aqueous solution of the same species. Clearly, a 22 nm red shift as well as a 38 nm band broadening (FWHM = 39 and 77 nm in deionized water and on glass, respectively) of the emission spectra are observed upon surface deposition.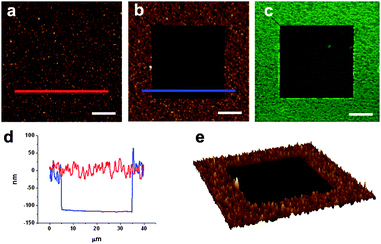 | ||
| Fig. 1 (a) AFM image of a fluorescein film deposited by spin-coating on glass. (b) AFM image of the same area after scratching a 30 × 30 μm2 region. (c) Corresponding confocal fluorescence image. (d) Height profiles taken from (b) showing the corrugation of the fluorescein film (red line) and the thickness of the film removed by scratching (blue line). Finally an AFM 3D image of the scratched area is shown in (e). Scale bars are 10 μm. | ||
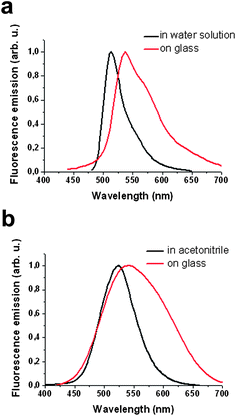 | ||
| Fig. 2 (a) Emission spectra of the dianionic form of compound 1 in aqueous solution (black line) and upon deposition on a glass slide (red line). (b) Emission spectra of the anionic form of compound 2 in acetonitrile solution (black line) and upon deposition on a glass slide (red line). | ||
Similar results were obtained for spin-coated thin-films of compound 2 on glass, which was deposited in its most fluorescent anionic form from acetonitrile solution. AFM scratching experiments revealed a layer thickness of ∼95 nm, in agreement to that previously described for compound 1.
Analogously, the fluorescence spectrum of a2 on glass also experiences a red-shift (∼20 nm) and broadens (from FWHM = 68 to 130 nm) with respect to that measured in acetonitrile (see Fig. 2b). Therefore, the spectral range at which thin-films of compounds 1 and 2 can be investigated considerably enlarges upon deposition on glass.
Self-organization versus stamp-assisted deposition
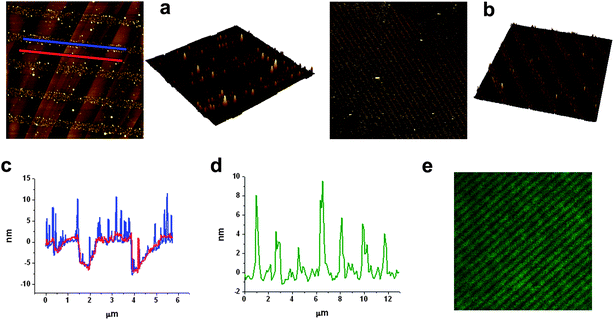
Surface deposition of compounds 1 and 2 was studied on two different substrates: highly ordered pyrolytic graphite (HOPG) and glass. Glass exhibits favorable properties for fluorophore deposition since it is chemically inert, optically transparent and provides good electrical insulation. On the other side, HOPG was selected for comparison purposes. HOPG is atomically flat (the glass substrates used present roughness values up to 2 nm) and it exhibits preferential directions that can induce anisotropy on the molecular structures. Moreover, it is more hydrophobic, which allowed us to study the influence of the hydrophobic/hydrophilic character of the substrate on the molecular organization. Drop-casting of compound 2 was attempted from dichloromethane and octanol solutions with concentrations ranging from 10−1 mM to 10−5 mM. Both solvents exhibit very different boiling points (40° for dichloromethane and 195° for octanol) leading to different evaporation times, and most likely, to different reorganization mechanisms for the same molecule. Moreover, octanol can participate in the formation of hydrogen-bonds with the catechol group. In the case of compound 1, an ethanol solution was used for the drop casting. Self-organization processes were studied by AFM immediately after solvent evaporation of drop-casted solutions of both compounds. Such studies were done at least over three different areas, to assess the reproducibility of the results. | ||
| Fig. 3 AFM images of 2 deposited by drop-casting on: (a) HOPG from a 0.1 mM dichloromethane solution; (b) HOPG from a 10−5 mM dichloromethane solution; (c) HOPG from a 0.1 mM octanol solution; (d) glass from a 0.1 mM acetonitrile solution. AFM images of 1 deposited by drop-casting on: (e) HOPG from a 0.1 mM ethanol solution; (f) glass from a 0.1 mM ethanol solution. Scale bars are 2 μm (a), 0.4 μm (b), 0.2 μm (c), 1.5 μm (d), 1 μm (e) and 10 μm (f). | ||
As expected, structuration of compounds 1 and 2 on glass exhibited a completely different behavior. Whereas drop-casting of compounds 1 and 2 on HOPG resulted in the formation of well-defined structures, deposition on glass originated the formation of more or less homogeneous films lacking any defined structural feature. This fact is mostly likely due to its higher hydrophilic character and therefore to its higher interaction with polar compounds 1 and 2; i.e. due to its better wettability. Deposition of 1 from an ethanol solution resulted in the formation of a thin film with a considerable corrugation ranging from 25 to 75 nm. Drop casting deposition of compound 2 from an acetonitrile solution resulted on the formation of a discontinuous film with profile heights that ranges from 4 nm to 6 nm. Interestingly, small-angle X-ray scattering (SAXS) experiments revealed the formation of amorphous layers lacking any order in both cases. As discussed below, this would favor the sensing activity of arrays of 1 and 2 on glass, since their amorphous structure should facilitate the diffusion of the analytes of interest through the chemosensing layer.
 | ||
| Fig. 4 AFM and fluorescence images of 2 deposited by LCW: (a) 8 × 8 μm2 AFM image of 2 deposited on HOPG from an octanol solution (inset is a 3D image of a zoomed area) (b) 50 × 50 μm2 AFM image of 2 deposited on glass from an octanol solution (inset is a 3D image of a zoomed area); (c) height profile of the HOPG steps in an area patterned with compound 2 (blue line) and in an unpatterned area (red line); (d) height profile of the LCW patterned stripes of 2 in (b). (e) 31 × 31 μm2 confocal fluorescence image of compound 2 on glass. | ||
In our case, stamps with different dimensions and features were successfully used. The resulting molecular arrays were characterized by AFM topography as well as by fluorescence microscopy. As an example, 5 × 5 μm2 and 20 nm high square motives of 1 are shown in Fig. 5a and 5b. Other motives can be distinguished as well. For instance, 20 × 20 μm2 square-networks with heights between 15–35 nm are shown in Fig. 5c. Finally the fluorescence image of a 220 × 220 μm2 area of the negative replica of 5 × 5 μm2 square-networks is given in Fig. 5d. These results nicely confirm the viability of μCP to prepare nanostructured arrays of compounds 1 and 2, as previously observed for LCW.
 | ||
| Fig. 5 (a) 50 × 50 μm2 3D-AFM image and (b) 35 × 35 μm2 fluorescence image of the same area of microstructures of compound 1 deposited on glass by μCP. (c) 80 × 80 μm2 AFM image of a negative replica of μCP-imprinted large squares and (d) 222 × 222 μm2 fluorescence image of a similar replica motive of 1 on glass. | ||
Acid–base studies
The multistate acid–base equilibria of compounds 1 and 2 (see Scheme 1) and their corresponding fluorescence switching-like behavior have already been established in solution.20,21 Herein we thoroughly investigated their pH-induced interconversion on structured arrays physisorbed on glass. With this aim, the different motives previously obtained by LCW and μCP were exposed to atmospheres (gas flow) of different acidities: HCl–N2 (pH ∼ 2), acetic acid–N2 (pH ∼ 4), air (pH ∼ 6) and triethylamine–N2 (pH ∼ 12). According to their pKa values, the formation of distinct states of 1 and 2 with different emission properties is favored in each case, and the pH-induced interconversion between them monitored by confocal fluorescence microscopy.The trend observed for all the structured arrays is consistent with that already described in solution. In the case of compound 1, the non-fluorescent c1 and n1 states (ϕf ∼ 0) were mainly formed at pH∼2, accounting for the low intensity of the measured emission. An increase to pH∼6 led to the formation of the monoanionic a1 state (ϕf = 0.37), thus resulting in a significant emission increase. Finally, a further pH increase up to pH∼12 gave rise to the formation of the highly-fluorescent dianionic d1 state (ϕf = 0.93) and to a maximum emission value. A continuous increase of fluorescence emission with pH was also observed for the arrays of 2. This behaviour arises from both the particular optical properties of this compound and the experimental excitation and detection conditions used in our fluorescence microscopy measurements. At those conditions only the anionic a2 state of this species appears to be fluorescent, thus accounting for the pH-dependence observed for the emission arising from nanostructured samples of 2; i.e. low, intermediate and high fluorescence intensities being measured at pH ∼ 2, 6 and 12, respectively.
As an example, Fig. 6 depicts the fluorescence images of a 20 × 20 μm2 LCW-structured area of 1 sequentially exposed to different acidity flows. The array consists of ∼800 nm width parallel lines that were found to be slightly fluorescent in air (Fig. 6b). Under these experimental conditions the molecule was mainly in its monoanionic a1 state, as confirmed by Raman spectroscopy. The spectrum showed peaks at 1185, 1331, 1418, 1556 and 1598 cm−1 similar to those found for bulk solution measurements.24 Then, exposure of the initial array to a HCl–N2 (pH ∼ 2) gas flow induced the formation of the non-fluorescent n1 state, as shown in Fig. 6a. On the contrary, exposure to a triethylamine–N2 (pH ∼ 12) flow formed the highly fluorescent d1 species as confirmed by Raman spectroscopy (peaks at 1170 and 1309 cm−1) (Fig. 6c). As a consequence, the intensity profiles in Fig. 6d show a dramatic increase of the fluorescence emission of LCW-imprinted lines of 1 upon pH increase. Thus, an average maximum fluorescence intensity of 12, 55 and 141 kcount s−1 was measured at pH ∼ 2, 6 and 12, respectively.
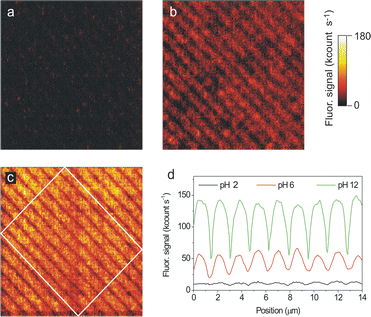 | ||
| Fig. 6 Consecutive fluorescence images of a 20 × 20 μm2 area of a sample prepared by LCW deposition of 1 on glass from an ethanol solution (a) after exposure to an acidic gas flow (HCl–N2), (b) after exposure to air and (c) after exposure to a basic gas flow (triethylamine–N2). All images were obtained with the same instrument settings (λexc = 532 nm, power density ∼0.25 kW cm−2, pixel rate = 1 kHz). (d) Mean intensity cross sections of the fluorescence images. The profiles plotted are obtained by averaging the signal within the region confined inside the white rectangle in image c). | ||
In all the cases the fluorescence changes observed upon pH variation were instantaneous on the time-scale of our experiments, i.e. on the experimental time required to change the acidity of the medium (less than 1 s). Therefore, this suggests that a very fast diffusion of the gas through the physisorbed molecules takes place thanks to its amorphous character (see previous sections). It is also important to emphasize that the sensitivity of the nanostructured samples is higher than that found for the corresponding thin-films. Such a sensitivity increase comes from comparison of the responses found for the functionalized and non-functionalized areas. For instance, a fluorescence intensity increase FpH=12/FpH=2 of 23 was determined for the LCW nanometre-sized imprinted stripes in Fig. 6, while a significantly lower value FpH–12/FpH–2 of 13 was found for a thin-film. Such considerable variation comes from the background correction applied to the nanostructured sample. This represents another clear advantage of nanopatterning functional surfaces for chemosensing purposes.
Structuration by covalent bonding
The fluorescent structures previously described have been shown to exhibit high-sensitivity and fast response time though their use is restricted to gas flow detection. As expected, direct exposure to liquid samples was found to destroy the sample. To demonstrate the viability of our approach in other media we have also fabricated arrays of a compound 1 derivative that can be chemically attached to a solid support. Initially, a glass slide was functionalized with amino alkylsilanes using a well-described silanization methodology.25 Afterwards, a commercial amino-reactive derivative of 1 (fluorescein isothiocyanate, fitc) was imprinted by μCP onto the pre-treated surface. Reaction of the isothiocyanate with the amino groups of the functionalized surface led to the formation of a molecular monolayer. Moreover, extensive rinsing was done to ensure the complete removal of all remaining physisorbed molecules. The resulting sample was inspected by fluorescence and atomic force microscopies and the results are shown in Fig. 7. The fluorescence image of a 170 × 130 μm2 area in Fig. 7a reveals the effective covalent patterning of fitc in the form of dots of 5 μm in diameter. The height of the patterned dots measured by AFM (≤1 nm, see Fig. 7b) is consistent with a monolayer of fitc molecules being printed onto the surface. Such studies were done at least over three different areas, to assess the reproducibility of the results.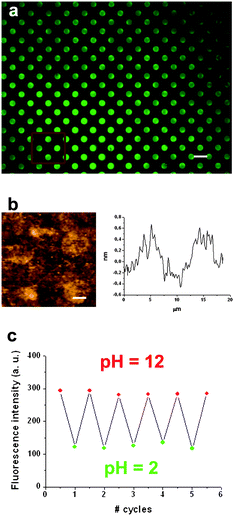 | ||
| Fig. 7 (a) Fluorescence image of a 170 × 130 μm2 area of multiple dots of fitc of 5 μm in diameter deposited on a functionalized glass substrate by μCP. Scale bar is 10 μm. (b) Contact AFM image and height profile of a 2 × 2 array of dots of the same sample. Scale bar is 2.5 μm. In (c), the reversible fluorescence emission response of the fitc array upon consecutive cycles of pH variation is shown, for which the sample was immersed into buffered solutions of pH 2 and pH 12. The fluorescence intensities plotted were obtained by averaging the signal of the same dots each time and were background-corrected. | ||
As previously described for the fluorescein physisorbed nanostructures, a continuous increase of fluorescence emission with pH was observed for the ftic imprinted dots onto glass. Indeed, the background-corrected average intensities measured were 115, 260 and 295 kcount s−1 at pH = 2 (neutral), 6 (monoanionic) and 12 (dianionic), respectively. This represents a fluorescence intensity increase of FpH–12/FpH–2 = 2.6, considerably smaller than those obtained for the physisorbed nanostructures or thin-films of compound 1. This fact is most likely due to the differential influence of the surface and intermolecular interactions on the emission properties of 1 upon direct deposition or covalent bonding. Indeed, the spectrum of physisorbed compound 1 on glass was found to be 8 nm broader and 7 nm red-shifted in comparison with the spectrum of the covalently-attached fitc (the opposite behavior is found in solution, where fitc is 5 nm red-shifted in comparison with compound 1). This clearly demonstrates that the molecule–substrate and intermolecular interactions found upon deposition onto a surface may significantly vary the optical properties of a fluorescent chemosensor and therefore, its sensing capabilities.
Finally, to evaluate the robustness and reproducibility of the changes experienced by covalently-bonded structures of fitc upon pH variations, five consecutive exposures (cycles) to aqueous acid and base solutions were done, while simultaneously measuring their fluorescence response. Fig. 7b displays the average emission intensities retrieved for the dots of fitc shown in Fig. 7a. Clearly, repetitive values of emission intensity were obtained at both pH = 2 and pH = 12, thus indicating the lack of degradation of the analyzed surface and its potential applicability to undertake reproducible and sensitive pH measurements.
Conclusions
Arrays of fluorescent pH-responsive multistate molecules on surfaces have been fabricated by means of two different soft lithographic techniques, LCW and μCP. Direct physisorption onto the desired surface can be attained in a very simple manner, without the need for tedious modification of the molecular systems neither of the supporting platforms. However, the arrays lack of the stability required to be used with liquids. This situation is solved by the use of covalent bonding though this requires a previous functionalization of both the molecule and the surface. Interestingly, in both cases their acid–base switching behavior remains once structured on a surface with small variations of their emission spectrum. In this way, the patterns generated have shown fluorescence sensing capabilities when they are exposed to atmospheres of different acidity over large pH-windows. These results open the door to the development of novel molecular-based sensing capabilities thanks to their fast responses.Experimental
Materials
Compound 2 was synthesized as previously reported.21 Fluorescein was purchased from Fluka. FITC (Fluorescein Isothiocyanate) was purchased from Invitrogen. PDMS (Polydimethylsiloxane) Sylgard R 184 was purchased from Dow Corning Corporation. Triethylamine and acetic acid were purchased from Panreac. All the rest of reagents used were purchased from Aldrich, and all of them were used as received.Atomic Force Microscopy (AFM)
All AFM images were recorded on an Agilent 5500 AFM/SPM microscope (formerly Molecular Imaging PicoPlus AFM). The AFM was operated in semi-contact (“acoustic”) mode, except for the measurements in Fig. 7b (contact mode). A multi-purpose low-coherence scanner with scan range up to 90 μm was used for imaging samples under ambient conditions. The AFM probes were type NCH silicon pointprobes (force constant ≈ 42 N m−1, resonant frequency ≈ 330 KHz) from Nanosensors (Neuchatel, Switzerland). The scan rate was tuned proportionally to the area scanned and was kept in the 0.5–1.2 Hz range. The typical value of drive amplitude was 1.0–1.4 V. The resolution of image acquisition was 512 pixels per line. AFM image processing and rendering was done with the Gwyddion 2.10 software.Micro Contact Printing (μCP)
PDMS replicas were produced by pouring a precursor mixture over a pre-structured silicon wafer (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, curing agent to prepolymer, Sylgard 184, Dow Corning, US), which previously was vigorously stirred and degassed in vacuum, 30 min. It was then cured in an oven for 24 h at ambient pressure and 80°. The thickness of the PDMS stamp produced was between 2 and 3 mm.
:10, curing agent to prepolymer, Sylgard 184, Dow Corning, US), which previously was vigorously stirred and degassed in vacuum, 30 min. It was then cured in an oven for 24 h at ambient pressure and 80°. The thickness of the PDMS stamp produced was between 2 and 3 mm.
Optical microscopy
Fluorescence optical images were obtained with a Zeiss Axio Observer Z-1 inverted microscope with motorized XY stage, Hg lamp excitation source and standard filters. 20×, 50×, and 100× air objectives from Zeiss were used in these studies.A custom-made confocal scanning fluorescence microscope with a homebuilt gas chamber was used for acid/base in situ fluorescence studies, as explained elsewhere.17 This instrument consisted of a high-numerical-aperture (NA) oil-immersion objective (Olympus, 60×, NA = 1.42) mounted on an inverted focusing unit (Olympus, BXFM), with which the circularly polarized light from a continuous green diode laser (λ = 532 nm, Z-laser, Z20RG) was focused onto the sample. The resulting fluorescence emission was collected by the same objective, spectrally filtered from excitation light using dichroic (Omega, 550DRLP) and longpass (Omega, 550ALP) filters, and finally detected in an avalanche photodiode (Perkin-Elmer, SPCM-AQR-14). Fluorescence images were obtained by raster-scanning micrometre-sized areas of the sample by means of a piezoscanner with position feedback control (Physik Instrumente, P-710) at 1-kHz pixel rate and with a laser excitation power density within the range 50–500 W cm−2. The movement of the scanner as well as the collection of the signal for the photodetector was controlled by means of a LabVIEW (National Instruments) program. To induce deprotonation–protonation of fluorescein and compound 1 on the surface during the confocal fluorescence measurements, the sample was exposed to triethylamine–N2, air or acetic acid–N2 flows in the interior of a homebuilt gas-chamber. The same chamber was used for measurements under liquid, in which different buffer solutions were used.
A commercial confocal microscope (Leica TCS SP2 AOBS) with blue diode laser excitation (λexc = 405 nm) was also used to measure the fluorescence emission spectra of the compounds.
Acknowledgements
We acknowledge the financial support of the Ministerio de Educación y Ciencia (MEC) through projects MAT2009-13977-C03-03, CTQ2007-60613/BQU and CTQ2009-07469. A. M.-O. thanks the Generalitat de Catalunya for his predoctoral grant. We also would like to thank Dr Jordi Esquena for SAXS measurements and Dr Francesc Alsina for Raman measurements.References
- L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993 RSC and references therein; O. S. Wolfbeis, J. Mater. Chem., 2005, 15, 2657 Search PubMed; D. R. Walt, Curr. Opin. Chem. Biol., 2002, 6, 689 RSC; R. B. H. Veenhuis, E. J. van der Wouden, J. W. Van Nieuwkasteele, A. Van der Berg and J. C. T. Eijkel, Lab Chip, 2009, 9, 3472 CrossRef CAS.
- A. Schiller, R. A. Wessling and B. Singaram, Angew. Chem., Int. Ed., 2007, 46, 6457 CrossRef CAS; J. F. Folmer-Andersen, M. Kitamura and E. V. Anslyn, J. Am. Chem. Soc., 2006, 128, 5652 CrossRef CAS; H. Zhou, L. Baldini, J. Hong, A. J. Wilson and A. D. Hamilton, J. Am. Chem. Soc., 2006, 128, 2421 CrossRef CAS; R. Jelinek and S. Kolusheva, Chem. Rev., 2004, 104, 5987 CrossRef CAS; A. Buryak and K. Severin, Angew. Chem., Int. Ed., 2005, 44, 7935 CrossRef CAS.
- M. Crego-Calama and D. N. Reinhoudt, Adv. Mater., 2001, 13, 1171 CrossRef CAS; Y. Zheng, J. Orbulescu, X. Ji, F. M. Andreopoulos, S. M. Pham and R. M. Leblanc, J. Am. Chem. Soc., 2003, 125, 2680 CrossRef CAS; L. Basabe-Desmonts, F. van der Baan, R. S. Zimmerman, D. N. Reinhoudt and M. Crego-Calama, Sensors, 2007, 7, 1731 CrossRef CAS.
- N. A. Rakow and K. S. Suslick, Nature, 2000, 406, 710 CrossRef CAS; J. S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864 CrossRef CAS; Y. Liu, R. C. Mills, J. M. Boncella and K. S. Schanze, Langmuir, 2001, 17, 7452 CrossRef CAS; K. J. Albert and D. R. Walt, Anal. Chem., 2000, 72, 1947 CrossRef CAS; T. A. Dickinson, J. White, J. S. Kauer and D. R. Walt, Nature, 1996, 382, 697 CrossRef CAS.
- U. Kosch, I. Klimant, T. Werner and O. S. Wolfbeis, Anal. Chem., 1998, 70, 3892 CrossRef CAS; P. Mela, S. Onclin, M. H. Goedbloed, S. Levi, M. F. García-Parajó, N. F. van Hulst, B. J. Ravoo, D. N. Reinhoudt and A. van den Berg, Lab Chip, 2005, 5, 163 RSC.
- K. Schult, A. Katerkamp, D. Trau, F. Grawe, K. Cammann and M. Meusel, Anal. Chem., 1999, 71, 5430 CrossRef CAS.
- For a recent review on fiber-optic pH sensors see: O. S. Wofbeis, Anal. Chem., 2008, 80, 4269 Search PubMed.
- J. Fritz, Analyst, 2008, 133, 855 RSC; M. Watari, J. Galbraith, H.-P. Lang, M. Sousa, M. Hegner, C. Gerber, M. A. Horton and R. A. McKendry, J. Am. Chem. Soc., 2007, 129, 601 CrossRef CAS.
- A. S. Vasylevska, A. A. Karasyov, S. M. Borisov and C. Krause, Anal. Bioanal. Chem., 2007, 387, 2131 CrossRef CAS; A. P. de Silva, S. S. K. de Silva, N. C. W. Goonesekera, H. Q. N. Gunaratne, P. L. Lynch, R. K. Nesbitt, S. T. Patuwathavithana and N. L. D. Ramyalai, J. Am. Chem. Soc., 2007, 129, 3050 CrossRef.
- S. Lenhert, P. Sun, Y. Wang, H. Fuchs and C. A. Mirkin, Small, 2007, 3, 71 CrossRef CAS; S. Sekula, J. Fuchs, S. Weg-Renners, P. Nagel, S. Schuppler, J. Fragala, N. Theilacker, M. Franzreb, C. Wingren, P. Ellmark, C. A. K. Borrebaeck, C. A. Mirkin, H. Fuchs and S. Lenhert, Small, 2008, 4, 1785 CrossRef CAS.
- T. Gunnlaugsson and D. Parker, Chem. Commun., 1998, 511 RSC; M. Su, H. Ma, Q. Ma, Z. Wang, J. Yang and M. Wang, Chem. Commun., 2001, 960 RSC; F. Pina, J. C. Lima, C. Lodeiro, J. S. de Melo, P. Díaz, M. T. Albelda and E. García-España, J. Phys. Chem. A, 2002, 106, 8207 CrossRef CAS; V. F. Valuk, G. Duportail and V. G. Pivovarenko, J. Photochem. Photobiol., A, 2005, 175, 226 CrossRef CAS; G. Nishimura, Y. Shiraishi and T. Hirai, Chem. Commun., 2005, 5313 RSC; B. Tang, X. Liu, K. Xu, H. Huang, G. Yang and L. An, Chem. Commun., 2007, 3726 RSC.
- P. Pallavicini, Y. A. Diaz-Fernandez and L. Pasotti, Analyst, 2009, 134, 2147 RSC; Y. Shiraishi, Y. Tokitoh, G. Nishimura and T. Hirai, J. Phys. Chem. B, 2007, 111, 5090 CrossRef CAS; A. P. de Silva, H. Q. N. Gunaratne and C. P. McCoy, Chem. Commun., 1996, 2399 RSC; T. Gunnlaugsson, J. P. Leonard, K. Sénéchal and A. J. Harte, J. Am. Chem. Soc., 2003, 125, 12062 CrossRef CAS; S. A. de Silva, K. C. Loo, B. Amorelli, S. L. Pathirana, M. Nyakirangáni, M. Dharmasena, S. Demarais, B. Dorcley, P. Pullay and Y. A. Salih, J. Mater. Chem., 2005, 15, 2791 RSC; Y. Diaz-Fernandez, F. Fori, C. Mangano, P. Pallavicini, S. Patroni, A. Perez-Gramatges and S. Rodriguez-Calvo, Chem.–Eur. J., 2006, 12, 921 CrossRef CAS.
- P. Pallavicini, V. Amendola, C. Massera, E. Mundum and A. Taglietti, Chem. Commun., 2002, 2452 RSC; V. Amendola, L. Fabrrizzi, C. Mangano, H. Miller, P. Pallavicini, A. Parotti and A. Taglietti, Angew. Chem., 2002, 114, 2665 CrossRef; V. Amendola, L. Fabrrizzi, C. Mangano, H. Miller, P. Pallavicini, A. Parotti and A. Taglietti, Angew. Chem., Int. Ed., 2002, 41, 2553 CrossRef CAS; Z. Wang, G. Zheng and P. Lu, Org. Lett., 2005, 7, 3669 CrossRef CAS; G. Zheng, Z. Wang, L. Tang, P. Lu and W. P. Weber, Sens. Actuators, B, 2007, 122, 389 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 2nd edn, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- R. Sjöback, J. Nygren and M. Kubista, Spectrochim. Acta, Part A, 1995, 51, L7 CrossRef.
- A. Martínez-Otero, J. Hernando, D. Ruiz-Molina and D. Maspoch, Small, 2008, 4, 2131 CrossRef CAS.
- A preliminary communication of this work has already been published: A. Martínez-Otero, E. Evangelio, R. Alibés, J. L. Bourdelande, D. Ruiz-Molina, F. Busque and J. Hernando, Langmuir, 2008, 24, 2963 Search PubMed.
- (a) M. Cavallini, M. Facchini, M. Massi and F. Biscarini, Synth. Met., 2004, 146, 283 CrossRef CAS; (b) M. Cavallini, C. Albonetti and F. Biscarini, Adv. Mater., 2009, 21, 1043 CrossRef CAS; (c) M. Cavallini, F. Biscarini, J. Gomez-Segura, D. Ruiz and J. Veciana, Nano Lett., 2003, 3, 1527 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550 CrossRef CAS.
- Previous examples of optically-active catechol families have already been reported: (a) Ch. Aronica, A. Venancio-Marques, J. Chauvin, V. Robert and G. Lemercier, Chem.–Eur. J., 2009, 15, 5047 CrossRef CAS; (b) G. Poneti, M. Mannini, L. Sorace, P. Sainctavit, M. A. Arrio, A. Rogalev, F. Wilhelm and A. Dei, ChemPhysChem, 2009, 10, 2090 CrossRef CAS; (c) P. Dapporto, A. Dei, G. Poneti and L. Sorace, Chem.–Eur. J., 2008, 14, 10915 CrossRef CAS.
- E. Evangelio, J. Hernando, I. Imaz, G. G. Bardaji, R. Alibes, F. Busque and D. Ruiz-Molina, Chem.–Eur. J., 2008, 14, 9754 CrossRef CAS.
- M. F. Choi, J. Photochem. Photobiol., A, 1998, 114, 235 CrossRef CAS; M. T. Charreyre, O. Tcherkasskaya, M. A. Winnik, A. Hiver, T. Delair, P. Cros, C. Pichot and B. Mandrand, Langmuir, 1997, 13, 3103 CrossRef CAS; A. K. Gaigalas, L. Wang, A. Schwartz, G. E. Marti and R. F. Vogt, J. Res. Natl. Stand. Technol., 2005, 110, 101 CAS.
- Y. D. Yan, T. Sun, Y. C. Liang and S. Dong, Int. J. Mach. Tools Manuf., 2007, 47, 1651 CrossRef.
- L. Wang, A. Roitberg, C. Meuse and A. K. Gaigalas, Spectrochim. Acta, Part A, 2001, 57, 1781 CrossRef CAS.
- N. K. Kamisetty, S. P. Pack, M. Nonogawa, K. C. Devarayapalli, T. Kodaki and K. Makino, Anal. Bioanal. Chem., 2006, 386, 1649 CrossRef CAS.
Footnote |
| † Dedicated to Prof. J. Font in honor of his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2010 |