A new CoFe2O4–Cr2O3–SiO2 fluorescent magnetic nanocomposite
Chandan
Borgohain
ab,
Kula Kamal
Senapati
ab,
Debabrata
Mishra
b,
Kanak Ch.
Sarma
c and
Prodeep
Phukan
*a
aDepartment of Chemistry, Gauhati University, Guwahati, 781014 Assam, India. E-mail: pphukan@yahoo.com; Fax: +91-361-2700311; Tel: +91-361-2570535
bIndian Institute of Technology Guwahati, Guwahati, 781039 Assam, India
cDepartment of Instrumentation and USIC, Gauhati University, Guwahati, 781014, Assam, India
First published on 10th September 2010
Abstract
A combined sonochemical co-precipitaion method has been developed for the synthesis of a CoFe2O4–Cr2O3–SiO2 magnetic nanocomposite. The synthesis involved the pre-synthesis of CoFe2O4–Cr2O3 nanoparticles, which were subsequently coated with SiO2 by treatment with tetraethyl orthosilicate. It was observed that the as-prepared CoFe2O4–Cr2O3–SiO2 nanocomposite exhibits photoluminescence properties without the addition of any external fluorescent marker. The fluorescent magnetic nanoparticles (FMNPs) had a typical diameter of 30 ± 5 nm and a saturation magnetization of 5.1 emu g-1 at room temperature. This as-prepared nanocomposite was used for staining cultured HeLa cells for fluorescence imaging.
Introduction
Magnetic nanoparticles have drawn significant attention in recent years from both a fundamental point of view and for applications in material science.1–4 Such particles are in high demand as new nanoscale technologies are beginning to change the scientific landscape in terms of medical diagnosis, treatment and prevention.5–7 Cobalt ferrite (CoFe2O4) is one of the most extensively studied ferrites and has been exploited for its potential utilization as an active component for high-density magnetic storage, spintronic devices and for the fabrication of sensors for biomedical applications and hyperthermia.8–10 Soler et al.11 have shown that CoFe2O4 has high structural stability and is reliable as a magnetic drug carrier in biological applications. Chromium oxide (Cr2O3) on the other hand is an important material since it has a high melting temperature, is resistance to oxidation and exhibits interesting optical, electrical and magnetic properties.12,13 Magnetically CoFe2O4–Cr2O3 is a two-phase exchanged coupled system consisting of a ferromagnet (CoFe2O4) biased by an antiferromagnet (Cr2O3) and such systems with ferro-antiferromagnetic coupling have been extensively studied in light of magnetoresistive read-head applications.14,15Recently, several types of nanoparticle have been used for bioanalysis. These include dye-doped nanoparticles,16 quantum dots (QDs),17 lanthanide (Ln3+) doped nanoparticles,18 magnetic nanoparticles19 and gold nanorods.20 These nanoparticles have their own unique properties and have been adapted for different applications in the field of bioanalysis. Compared with conventional organic dyes, luminescent nanoparticles are preferred as probes for bioapplications because of their photostability and strong luminescence. For instance, QD-integrated magnetic nanoparticles have been actively studied due to their excellent optical properties such as having narrow emission bands, continuous broad-band absorption and a high resistance to photobleaching in comparison to organic dyes. However the separation between the magnetic nanoparticles and the QDs is controlled using a layer-by-layer (LBL) approach21 to prevent quenching. Such nanoparticles are usually large (70–100 nm) affecting their stability. Moreover, this method uses reagents such as polyelectrolytes which are expensive.
For many applications such as magnetic recording and targeted drug delivery, particles with a higher magnetic moment and larger particle size are required. Furthermore it is difficult to produce non-agglomerated nanocrystals of ferromagnetic nanoparticles. Often larger particles (∼50 nm), just below the superparamagnetic threshold, are more suitable for applications such as targeted drug delivery as these parameters may influence drug loading, drug release, stability, toxicity and biological fate. Much effort has been made to synthesize cobalt ferrite with well-defined properties. These include important examples such as mechanochemical methods,22 sonochemical reactions,23 co-precipitation,24 micro-emulsion procedures,25 and others.26–32 One of the major disadvantages in most of these techniques is the lower degree of crystallinity in the resulting material, leading in turn to significant spin misalignment which reduces the net magnetic moment of the particle.
In this report we describe a method for the synthesis of a Cr2O3-doped CoFe2O4 nanoparticles coated with a monolayer of SiO2. A combined co-precipitation sonochemical technique has been developed in order to obtain highly crystalline magnetic nanoparticles of 30 ± 5 nm particle size. It was observed that this nanocomposite shows photoluminescence properties without the extra addition of any fluorescent marker. The use of magnetic fluid nanoparticles (MFNPs) was tested for bio-imaging human cervical cancer cells (HeLa). While no studies of such CoFe2O4–Cr2O3–SiO2 nanoparticles for in vitro and in vivo applications have been reported so far, such conjugates may been used as bioprobes in which the fluorescent part may be used as an effective tool for imaging biological cells while the magnetic part may be used as a therapeutic agent for performing hyperthermia treatment. The development of such a MFNP with properties for bio-imaging and for the transport of pharmaceuticals to specific sites in the body constitutes a powerful tool for gene/drug therapy.33
Experimental
The synthesis of the CoFe2O4–Cr2O3–SiO2 magnetic nanocomposite was achieved in three successive steps. Initially uncapped CoFe2O4 was synthesized which was then coated with Cr2O3. Subsequent treatment of the CoFe2O4–Cr2O3 nanoparticles with trietylorthosilicate produced the CoFe2O4–Cr2O3–SiO2 magnetic nanocomposite. The procedure is described below.Synthesis of CoFe2O4 nanoparticles
Two aqueous solutions of FeCl3 (1.5 g, 9.3 mmol, 50 mL) and CoCl2·6H2O (1 g, 4.2 mmol, 50 mL) were mixed in a 200 mL flat bottom flask and placed in an ultrasonic bath. An aqueous KOH solution (3 M, 25 mL) was added dropwise under an argon atmosphere with continuous ultrasonic irradiation (frequency 40 kHz at 40 kW). Prior to mixing, all these three solutions were sonicated for 30 min to remove dissolved oxygen. The temperature of the sonicator bath was raised up to 60 °C and the mixture was sonicated for a further 30 min in air. The reaction mixture was centrifuged (14![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 rpm) at ambient temperature for 15 min. The mixture was further subjected to successive sonication (30 min) and centrifugation (15 min) five times. The black precipitate was then separated, washed with copious amounts of distilled water followed by ethanol and kept overnight in an incubator at 60 °C for ageing. The precipitate was further dried in an oven at 100 °C for one hour and subsequently kept under high vacuum (10−2 bar) for one hour. Finally, the black particles were placed in 50 mL of dry ethanol and subjected to successive sonication (30 min) and centrifugation (15 min) repeatedly until a brown solution appeared. The precipitate was separated, dried and used for further modification.
000 rpm) at ambient temperature for 15 min. The mixture was further subjected to successive sonication (30 min) and centrifugation (15 min) five times. The black precipitate was then separated, washed with copious amounts of distilled water followed by ethanol and kept overnight in an incubator at 60 °C for ageing. The precipitate was further dried in an oven at 100 °C for one hour and subsequently kept under high vacuum (10−2 bar) for one hour. Finally, the black particles were placed in 50 mL of dry ethanol and subjected to successive sonication (30 min) and centrifugation (15 min) repeatedly until a brown solution appeared. The precipitate was separated, dried and used for further modification.
Synthesis of the CoFe2O4–Cr2O3 nanocomposite
The coating of a layer of Cr2O3 on the surface of CoFe2O4 nanoparticles was achieved by premixing a dispersion of CoFe2O4 nanoparticles in deionised water with appropriate molar ratios of Cr(OAc)3·H2O in a round-bottom flask in an ultrasonic bath. An aqueous solution of NaOH was added to the mixture in the presence of ultrasonic irradiation (frequency 40 kHz at 40 kW). Prior to mixing, all these four solutions were degassed by sonication for 30 min. The temperature of the sonicator bath was raised to 60 °C and the mixture was sonicated for a further 30 min in air. Brown precipitate formation was observed during this time. The reaction mixture was centrifuged (10000 rpm) at ambient temperature for 15 min. The brown precipitate was then separated, washed with copious amounts of distilled water followed by ethanol and kept overnight in an incubator at 60 °C for ageing. The precipitate was further dried in an oven at 100 °C for one hour and subsequently kept under high vacuum (10−2 bar) for one hour. Finally, the particles were placed in 50 mL of dry ethanol and subjected to successive sonication (30 min) and centrifugation (15 min) repeatedly until a brown solution appeared. The precipitate was separated, dried and held at 1000 °C in a muffle furnace for 10 hours to obtain a fine black powder. Energy-dispersive X-ray spectroscopy (EDX) analysis at this point showed excellent agreement between expected and observed values of the constituent elements which therefore confirmed the formation of CoFe2O4–Cr2O3.
Surface modification of CoFe2O4–Cr2O3 nanoparticles
The third synthesis step involves silica coating of the as-prepared CoFe2O4–Cr2O3 nanoparticle surfaces by the hydrolysis of tetraethyl orthosilicate (TEOS).34 The CoFe2O4–Cr2O3 nanoparticles (0.2 g) were dispersed in a mixture of ethanol (20 mL), water (9 mL) and ammonia (25%, 0.5 mL) under ultrasonication and then TEOS (0.5 mL) was added to the mixture. After three hours, the precipitate was isolated by centrifugation, washed with ethanol and water several times and dried at 80 °C under vacuum for two hours.Cell labeling
In the cell-labeling process, the cells were cultured in a 25 cm2 glass culture vial and the culture medium was a mixture of DMEM (Dulbecco's modified Eagle's medium), 10% inactivated fetal bovine serum, 50 units mL−1 of penicillin, 40 mg mL−1 of streptomycin, and 0.3 mg mL−1 of L-glutamine. Cells were cultured at 37 °C in a humidified atmosphere that was supplied with 5% CO2. When the number of cells reached ∼105 cells per vial, the culture medium was removed, and a mixture of 0.2 mL of nanoparticles in PBS buffer (2 mg mL−1) was added to the culture vial. After incubation for 20 h at 35 °C in 5% CO2 , ∼95% air incubator, the culture vial was washed 16 times using PBS buffer and was subjected to transmission electron microscopy (TEM) and fluorescence imaging.Cell viability experiments
The antiproliferative assay was carried out using a standard methylthiazoltetrazolium bromide (MTT) assay. The cells (5 × 104 cells in each 100 µL medium well) were plated in 0.07% DMSO as a control in 96-well plates and were incubated for 24 h. At the end of the treatments, each well was treated with MTT (10 µL), and after incubation for two hours, the absorption at 570 nm was read with a microplate reader.Characterization
To investigate the formation of CoFe2O4–Cr2O3 nanocomposites, IR studies and X-ray diffraction patterns were recorded on a Perkin–Elmer RXI FT-IR spectrometer using KBr pellets and on a Bruker AXD D8 using Cu Kα radiation (λ = 1.54178 Å). The samples for TEM were prepared by drying an ethanol dispersion of the particles on a carbon-coated copper grid. The particles were imaged on a 200 kV, JEOL JEM2100 transmission electron microscope. Quantitative elemental analysis was carried out with an Oxford energy-dispersive X-ray spectrometer mounted on the transmission electron microscope. The magnetic properties of the as-prepared CoFe2O4, CoFe2O4–Cr2O3 and CoFe2O4–Cr2O3–SiO2 nanoparticles were investigated using a vibrating sample magnetometer (Lakeshore 7410). UV–visible spectra and fluorescent spectra of the nanoparticles were recorded using a Varian Cary50 biospectrophotometer and Edinburgh FSP920 Instruments respectively. Quantum yield evaluations were made by comparing the integrated intensity of the luminescence of the sample with a standard solution of quinine sulphate. The UV–visible absorbance spectrum of the reference solvent and the sample solution were noted at the excitation wavelength of 260 nm and 360 nm. Cell labeling experiments were conducted under fluorescence microscopy using a Carl Zeiss, LSM 510 meta confocal laser scanning microscope.Results and discussion
Structural and morphological analysis
To understand the mechanisms behind the magnetic properties, we carried out detailed magnetic and microstrucural studies. The fluorescent properties of the nanoparticles were studied using a time-resolved steady-state photoluminescence spectrometer. Pure magnetic nanoparticles however may not be very useful in practical applications as they are likely to form large aggregates and their magnetic properties would change or they may undergo rapid biodegradation when they are exposed to biological systems. To remove these drawbacks we coated the nanoparticles with a layer of SiO2.Fig. 1(a) shows the XRD pattern of the CoFe2O4–Cr2O3 nanocomposite. The diffraction peaks and relative intensities of the pattern match well with the cubic spinel structure of CoFe2O4 (JCPDS–International center diffraction data, PDF cards 3-864 and 22-1086) and of Cr2O3 (JCPDS–International center diffraction data, PDF cards 06-0504).
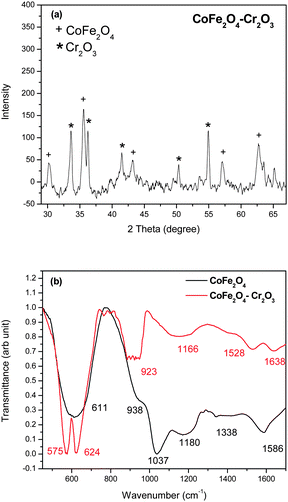 | ||
| Fig. 1 (a) XRD pattern and (b) FT-IR spectra of the as-prepared CoFe2O4–Cr2O3 particles. | ||
The crystallite size (D) of the nanoparticles were determined using the Scherrer formulae35 on the (311) peak of CoFe2O4 and was found to be 30 nm. The FT-IR spectra of the material (Fig. 1b) showed peaks at 611, 938, 1037, 1180, 1338 and 1586 cm−1 corresponding to CoFe2O4 and peaks at 575, 624, 923, 1155, 1528 and 1638 cm−1 corresponding to CoFe2O4–Cr2O3. The peak obtained at 611 cm−1 in the CoFe2O4 samples is attributed to the Fe–O or the Co–O bond.36 In the CoFe2O4–Cr2O3 samples the peak at 575 cm−1 corresponds to the Cr–O bond.37
The structural composition and crystallinity of the cobalt ferrite nanoparticles was further examined using TEM. Fig. 2 shows the TEM image of the cobalt–ferrite nanocrystals deposited on a carbon-coated copper grid. A TEM image of the CoFe2O4–Cr2O3 nanoparticle is shown in Fig. 2a and Fig. 2b shows the TEM image of the CoFe2O4–Cr2O3–SiO2 nanocomposite. The SAED pattern (Fig. 2d) obtained from TEM showed CoFe2O4–Cr2O3–SiO2, with the rings corresponding to reflections from the planes of CoFe2O4, Cr2O3 and SiO2. The average size of the nanoparticles from the TEM analysis (Fig. 2a–b) was found to be 30 ± 5 nm.
 | ||
| Fig. 2 TEM images. (a) TEM image of CoFe2O4–Cr2O3,(b) TEM image of CoFe2O4–Cr2O3–SiO2 nanocomposite, (c) HRTEM images of CoFe2O4–Cr2O3–SiO2 nanocomposite and (d) SAED pattern of CoFe2O4–Cr2O3–SiO2 nanocomposite. | ||
Magnetic properties
It is well known that the magnetic properties of CoFe2O4 depend on the chemical nature of Co, since Fe3+ species are evenly distributed in the structure at tetrahedral and octahedral interstices and are antiferromagnetically coupled. Such coupling cancels the moment contribution from Fe3+ and hence the moment contribution is solely dependent on Co2+.38 Incorporation of antiferromagnetic Cr2O3 and SiO2 in the Co–Fe–O matrix may cause changes in the magnetic properties of the material. The Magnetisation–Hysteresis (M–H) loop was taken at room temperature with a maximum applied field of ± 2 T. From the hysteresis loop, both saturation magnetization, coercivity and retentivity values were extracted.Fig. 3 shows that the prepared CoFe2O4–Cr2O3–SiO2 particles are ferromagnetic at room temperature with a saturation magnetization of 5.1 emu gm−1 and coercivity of 482 Oe. It has recently been demonstrated by Jordan et al.39 that large coercivity magnetic nanoparticles have the ability to self-heat when irradiated by electromagnetic irradiation. This is known as magnetic fluid hyperthermia (MFH)40 and can act as a therapeutic agent by itself. It can be seen from Table 1 that incorporation of Cr2O3 and SiO2 into the Co–Fe–O matrix has very little effect on the coercivity, however the magnetization and the retentivity decrease rapidly. Therefore by controlling the quantity of Cr2O3 and SiO2 in the Co–Fe–O matrix, magnetic properties of the nanocomposite can be tailored to suite different biomedical applications.
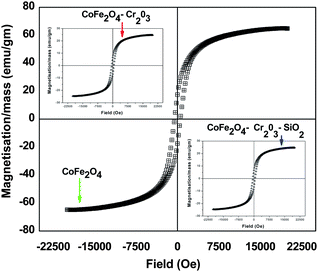 | ||
| Fig. 3 Hysteresis curves of CoFe2O4, CoFe2O4–Cr2O3 and CoFe2O4–Cr2O3–SiO2 nanocomposites. | ||
| Nanocomposite | Coercivity/Oe | Magnetization/mass emu gm−1 | Retentivity/emu gm−1 |
|---|---|---|---|
| CoFe2O4 | 539 | 64.97 | 17.49 |
| CoFe2O4–Cr2O3 | 529 | 24.74 | 6.62 |
| CoFe2O4–Cr2O3–SiO2 | 482 | 5.10 | 1.79 |
Spectral analysis
We have performed room-temperature optical absorption and photoluminescence to show the optical properties of the nanocomposite compared to its individual component (Cr2O3). Fig. 4 (a–d) shows the UV–visible absorption and fluorescence emission spectra of the CoFe2O4–Cr2O3–SiO2 particles. The sharp peak at 200 nm in the UV–visible spectra indicates that the colloids are well dispersed.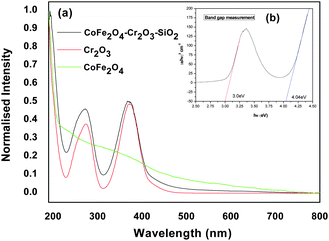 | ||
| Fig. 4 (a–b) Absorption spectra of CoFe2O4–Cr2O3–SiO2 at different concentrations, (c–d) absorbance of nanocomposites in DMEM over a period of 24 hrs. | ||
The UV–visible spectra of CoFe2O4–Cr2O3and Cr2O3 showed absorption bands at around 260 and 360 nm. The bandgap corresponding to the absorption bands was calculated by plotting (αhν)2vs.hν (Fig. 4b) using the relationship:41
| αhν = const (hν − Eg)n | (1) |
The value of α is obtained from the equation:42
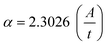 | (2) |
The extrapolation (Fig. 4b) of the straight line to α2 = 0 gives the value of the band-gap energy.
To further investigate absorption bands, the fluorescent properties of the nanoparticles were measured using a steady-state time-resolved spectrofluorometer at the wavelengths 260 and 360 nm for excitation scans and 350–600 nm for emission scans (Fig. 5). We did not observe any prominent fluorescence peaks for the as-prepared CoFe2O4–Cr2O3–SiO2 nanoparticles (Fig. 5a). However, when the sample was annealed at 1000 °C, a broad band at 460 nm appeared in the fluorescence spectrum, (Fig. 5b). This was attributed to lattice defects, such as interval atoms, displacement atoms and line defects resulting from grain boundary diffusion between CoFe2O4 and Cr2O3. The presence of defects in the nanocomposite is also supported by the HRTEM images of the sample (Fig. 2c). Such lattice defects are responsible for the luminescent properties of the nanocomposite.43
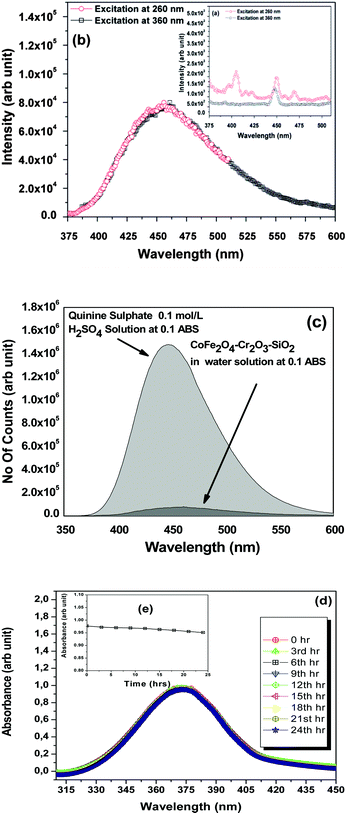 | ||
| Fig. 5 (a–b) Fluorescence emission spectra of CoFe2O4–Cr2O3–SiO2 particles excited at 260 and 360 nm; (c) emission spectra of quinine sulphate in 0.1 mol L−1 H2SO4 solution and CoFe2O4–Cr2O3–SiO2 dispersion in water at 0.1 ABS excited at 370 nm; (d–e) Absorbance of the nanocomposites in DMEM over a period of 24 h. | ||
The fluorescent efficiency of the nanocomposite was measured using quinine sulphate (ΦR = 0.55)44 in 0.1 mol L−1 H2SO4 solution as a standard using the relationship:45
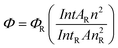 | (3) |
Where Φ is the quantum yield, Int is the area under the emission peak (on a wavelength scale), A is absorbance at the excitation wavelength of 370 nm, and n is the refractive index of the sample (we have taken the value of nR = 1.338,45n = 1.34639.46ΦR = 0.5547). The subscript R denotes the respective values of the reference substance.
The fluorescence spectrum of the solutions were recorded in a 20 mm fluorescence cuvette of constant slit width. In order to minimize reabsorption effects, absorbances at 0.1 above the excitation wavelength (at 370 nm) were used.48 The quantum-yield of the FMNP sample was found to be 0.0354 (Fig. 5c), which is appreciably good for bio-imaging.
The stability of the CoFe2O4–Cr2O3–SiO2 suspension was inferred from optical absorbance measurements taken at different intervals of time as studied by González-Caballero et al.49 The optical absorbance was studied at 360 nm as a function of time. We scanned the entire range of wavelengths from 310 nm to 450 nm for different intervals of time. All suspensions contained 0.1 g L−1 of CoFe2O4–Cr2O3–SiO2 particles in a mixture of DMEM, Sodium bicarbonate and water. Fig. 5(d–e) displayed the scanning spectra taken at 3 hour intervals up to 24 hours. From the spectra it was evident that there was no significant change in absorbance with time which revealed the stability of the suspension of the ferrite particles. However, the overall trend of A to decrease with time (ΔA = 0.04), due to magnetic interactions between the as-synthesized particles, demonstrates their stability.
Fluorescence imaging experiments
The combination of nanoscale dimensions, ferromagnetic properties and fluorescence for these bifunctional nanoparticles has prompted their use in medical imaging. Optical fluorescent techniques have high spatial resolution in cellular imaging and molecular event quantification. To demonstrate the utility of the fluorescent nanocomposite, we sought to apply it to cellular imaging. We have carried out our preliminary investigation using human cervical cancer cells (HeLa) with a mean cell diameter of 14.6 ± 0.8 µm as the model cell system and incorporated the MFNP in the cell.The as-synthesized SiO2-conjugated CoFe2O4–Cr2O3 nanoparticles were incubated with the HeLa cells, allowing an interaction on the cell surface resulting in the nanoparticles attached to the outer cell-membrane. The nanoparticles exhibited a surprisingly high level of cell internalization. While viewing the nanoparticle-labeled cells using TEM, we were able to follow the nanoparticles as they transported through the cell membrane (Fig. 6c) and into internalized compartments (Fig. 6a–b) confirmed by the SAED pattern (Fig 6d) taken of the cell compartments. The cellular uptake of the nanoparticles was further confirmed by the intercellular green fluorescence observed by morphology studies of the HeLa cells under fluorescence microscopy using a confocal laser scanning microscope (Fig 6e). We observed the morphological changes in the HeLa cells after treatment with the nanoparticles by fluorescence microscopic observation. The cells that were treated with the FMNPs did not show any cell shrinkage or rounded morphology. We observed a flattened morphology (Fig. 6e) of the cells, which shows the non-toxicity of the nanoparticles.50
 | ||
| Fig. 6 (a–c) TEM images of cultured HeLa cells; (d) SAED pattern taken of the internal compartment of the cells; (e–f) pattern and fluorescence image of cultured HeLa cells. | ||
Cell viability experiments
To evaluate the biocompatibility of FMNPs as bio-imaging probes, we investigated the cytotoxicity of FMNPs using a standard MTT assay.51 The cells (5 × 104 cells in 100 µL medium well−1) were plated in 0.07% DMSO media as a control in 96-well plates and were incubated for 24 h. After 24 h of growth, the medium was exchanged for the medium that contained the nanoparticles. The nanoparticle stock solution (150 µg mL−1) was prepared in water. From the stock solution, aliquots of nanoparticles were rapidly added to the culture medium. At the end of the treatments, each well was treated with MTT (10 µL), and after incubation for two hours, the absorption at 570 nm was read with a microplate reader. The cell viability was calculated using the following equation.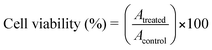 | (1) |
| Entry | A control | A treated | Cell viability | Average |
|---|---|---|---|---|
| 1 | 0.868 | 0.745 | 85.83 | — |
| 2 | 0.855 | 0.755 | 88.30 | — |
| 3 | 0.862 | 0.750 | 87.01 | 87.04 |
| 4 | 0.861 | 0.756 | 87.80 | — |
| 5 | 0.865 | 0.746 | 86.24 | — |
The FMNPs exhibited low toxicity (cell viability = 87%) towards HeLa cells even at a high concentration of 150 µg mL−1.
Conclusions
In conclusion, a combined sonochemical and co-precipitaion method has been developed for the synthesis of core–shell CoFe2O4–Cr2O3–SiO2 magnetic nanocomposites. The as-prepared CoFe2O4–Cr2O3–SiO2 nanocomposites exhibit photoluminescence properties without the addition of any external fluorescent marker. The fluorescent magnetic nanoparticles had a typical diameter of 30 ± 5 nm and a saturation magnetization of 5.1 emu g−1 at room temperature. The fluorescent nanocomposites were further utilized for staining the cultured HeLa cells for fluorescence imaging detection.Acknowledgements
Financial support from DST (India) for the TEM facility at CIF, IIT Guwahati (Grant No. SR/S5/NM-01/2005) and a Ramanna Fellowship to P. Phukan (Grant No. SR/S1/RFPC-07/2006) is gratefully acknowledged. The authors would also like to acknowledge the support from IIT Guwahati for the vibrating sample magnetometer, SEM and XRD facility. We thank reviewers for their valuable suggestions.References
- Y. Kitamoto, S. Kantake, S. Shirashaki, F. Abe and M. Naoe, J. Appl. Phys., 1999, 85, 4708 CrossRef CAS.
- M. Pardavi-Horvath, H. Montiel, G. Alvarez, I. Betancourt, R. Zamorano and R. Valenzuela, J. Magn. Magn. Mater., 2000, 171, 215.
- M. Uhlen, Nature, 1989, 340, 733 CrossRef CAS.
- D. G. Mitchell, J. Magn. Reson. Imaging, 1997, 7, 1 CrossRef CAS.
- Y. Okuhata, Adv. Drug Delivery Rev., 1999, 37, 121 CrossRef CAS.
- C. Bremer, V. Ntziachristos and R. Weissleder, Eur. Radiol., 2003, 13, 231.
- M. Doubrovin, I. Serganova, P. Mayer-Kuckuk, V. Ponomarev and R. G. Blasberg, Bioconjugate Chem., 2004, 15, 1376 CrossRef CAS.
- D. H. Han, H. L. Luo and Z. Yang, J. Magn. Magn. Mater., 1996, 161, 376 CrossRef CAS.
- K. Giri, E. M. Kirkpatrick, P. Moongkhamklang, S. A. Majetich and V. G. Harris, Appl. Phys. Lett., 2002, 80, 2341 CrossRef.
- H. R. Alexander Jr., T. S. Lawrence, S. A. Rosenberg, Cancer Principles and Practice of Oncology,Williams, & Wilkins, Philadelphia, 2008 Search PubMed.
- M. A. G. Soler, T. F. O. Melo, S. W. da Silva, E. C. D. Lima, A. C. M. Pimenta, V. K. Garg, A. C. Oliviera and P. C. Morais, J. Magn. Magn. Mater., 2004, 272–276, 2357 CrossRef CAS.
- R. H. Misho and W. A. Murad, Thin Solid Films, 1989, 169, 235 CrossRef CAS.
- Z. Pei and Y. Zhang, Mater. Lett., 2008, 62, 504 CrossRef CAS.
- C. Tsang, IEEE Trans. Magn., 1989, MAG-25, 3672.
- R. D. Hempstead, S. Krongelb and D. A. Thompson, IEEE Trans. Magn., 1978, 14, 521 CrossRef.
- S. Santra, P. Zhang, K. Wang, R. Tapec and W. Tan, Anal. Chem., 2001, 73, 4988–4993 CrossRef CAS.
- M. Bruchez Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976 RSC; D. K. Chatterjee, A. J. Rufaihah and Y. Zhang, Biomaterials, 2008, 29, 937 CrossRef CAS; S. A. Hilderbrand, F. Shao, C. Salthouse, U. Mahmood and R. Weissleder, Chem. Commun., 2009, 4188 RSC; S. Jiang, Y. Zhang, K. M. Lim, E. K. W. Sim and L. Ye, Nanotechnology, 2009, 20, 155101 CrossRef; M. Wang, C.-C. Mi, W.-X. Wang, C.-H. Liu, Y.-F. Wu, Z.-R. Xu, C.-B. Mao and S.-K. Xu, ACS Nano, 2009, 3, 1580 CrossRef CAS; T. Zako, H. Nagata, N. Terada, A. Utsumi, M. Sakono, M. Yohda, H. Ueda, K. Soga and M. Maeda, Biochem. Biophys. Res. Commun., 2009, 381, 54 CrossRef CAS; F. Vetrone, R. Naccache, A. J. de la Fuente, F. Sanz-Rodríguez, A. Blazquez-Castro, E. M. Rodriguez, D. Jaque, J. G. Solé and J. A. Capobianco, Nanoscale, 2010, 2, 495 RSC.
- R. Hergt, et al., J. Phys.: Condens. Matter, 2006, 18, S2919 CrossRef CAS.
- N. J. Durr, T. Larson, D. K. Smith, B. A. Korgel, K. Sokolov and A. Ben-Yakar, Nano Lett., 2007, 7, 941 CrossRef CAS.
- X. Hong, J. Li, M. J. Wang, J. J. Xu, W. Guo, J. H. Li, Y. B. Bai and T. J. Li, Chem. Mater., 2004, 16, 4022 CrossRef CAS.
- C. Jovalekić, M. Zdujic, A. Radakovic and M. Mitric, Mater. Lett., 1995, 24, 365 CrossRef CAS.
- K. V. P. M. Shafi, Y. Koltypin, A. Gedanken, R. Prozorov, J. Balogh, J. Lendvai and I. Felner, J. Phys. Chem. B, 1997, 101, 6409 CrossRef CAS.
- H. Tamura and E. Matijevic, J. Colloid Interface Sci., 1982, 90, 100 CAS.
- N. Moumen and M. P. Pileni, Chem. Mater., 1996, 8, 1128 CrossRef CAS.
- C. Xiangfeng, D. L. Jiang, Y. Guo and C. M. Zheng, Sens. Actuators, B, 2006, 120, 177 CrossRef.
- T. Sugimoto, Y. Shimotsuma and H. Itoh, Powder Technol., 1998, 85, 96.
- J. C. Hoh and I. I. Yaacob, J. Mater. Res., 2002, 17, 3105 CrossRef CAS.
- E. Manova, B. Kunev, D. Paneva, I. Mitov, L. Petrov, C. Estournes, C. D'Orléans, J.-H. Rehspringer and M. Kurmoo, Chem. Mater., 2004, 16, 5689 CrossRef CAS.
- T. Meron, Y. Rosenberg, Y. Lareah and G. Markovich, J. Magn. Magn. Mater., 2005, 292, 11 CrossRef CAS.
- E. Tirosh, G. Shemer and G. Markovich, Chem. Mater., 2006, 18, 465 CrossRef CAS.
- T. Hyeon, Y. Chung, J. Park, S. S. Lee, Y.-W. Kim and B. H. Park, J. Phys. Chem. B, 2002, 106, 6831 CrossRef CAS.
- J. P. Zimmer, S. W. Kim, S. Ohnishi, E. Tanaka, J. V. Frangioni and M. G. Bawendi, J. Am. Chem. Soc., 2006, 128, 2526 CrossRef CAS.
- M. Yu, J. Lin and J. Fang, Chem. Mater., 2005, 17, 1783 CrossRef CAS.
- B. D. Cullity, Elements of X-ray diffraction, Addison-Wesley Publishing Co. Inc., Reading, MA, 1978, 363 Search PubMed.
- V. M. Limaye, S. B. Singh, S. K. Date, D. Kothari, V. R. Reddy, A. Gupta, V. Sathe, R. J. Choudhury and S. K. Kulkarni, J. Phys. Chem. B, 2009, 113, 9070 CrossRef.
- M. Ocaña, J. Eur. Ceram. Soc., 2001, 21, 931 CrossRef CAS.
- F. Nakagomi, S. W. da Silva, V. K. Garg, A. C. Oliveira and P. C. Morais, J. Appl. Phys., 2007, 101, 09M514 CrossRef.
- A. Jordan, R. Scholz, P. Wust, H. Fahling and R. Felix, J. Magn. Magn. Mater., 1999, 201, 413 CrossRef CAS.
- R. E. Rosensweig, J. Magn. Magn. Mater., 2002, 252, 370 CrossRef CAS.
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354 CrossRef CAS.
- J. H. Park, J. Y. Kim, B. D. Chin, Y. C. Kim and O. O. Park, Nanotechnology, 2004, 15, 1217 CrossRef CAS.
- G. Zhang, W. Xu, Z. Li, W. Hu and Y. Wang, J. Magn. Magn. Mater., 2009, 321, 1424 CrossRef CAS.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107 CrossRef CAS.
- M. S. Attia, M. M. H. Khalil, A. A. Abdel-Shafi, G. M. Attia, S. Failla, G. Consiglio, P. Finocchiaro and M. S. A. Abdel-Mottaleb, Int. J. Photoenergy, 2007, 12530.
- M. Daimon and A. Masumura, Appl. Opt., 2007, 46, 3811 CrossRef.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229 CrossRef CAS.
- S. Dhami, A. J. de Mello, G. Rumbles, S. M. Bishop, D. Phillips and A. Beeby, Photochem. Photobiol., 1995, 61, 341 CrossRef CAS.
- J. de Vicente, A. V. Delgado, R. C. Plaza, J. D. G. Durán and F. González-Caballero, Langmuir, 2000, 16, 7954 CrossRef CAS.
- A. Sahu, N. Kasoju and U. Bora, Biomacromolecules, 2008, 9, 2905 CrossRef CAS.
- J. Yang, E.-K. Lim, H. J. Lee, J. Park, S. C. Lee, K. Lee, H.-G. Yoon, J.-S. Suh, Y.-M. Huh and S. Haam, Biomaterials, 2008, 29, 2548 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |