Functionalized graphene and graphene oxide solution via polyacrylate coating†
Arindam
Saha
a,
SK
Basiruddin
a,
S. C.
Ray
*b,
S. S.
Roy
c and
Nikhil R.
Jana
*a
aCentre for Advanced Materials, Indian Association for the Cultivation of Science, Kolkata, 700032, India. E-mail: camnrj@iacs.res.in
bSchool of Physics, University of the Witwatersrand, Johannesburg, South Africa. E-mail: Sekhar.Ray@wits.ac.za
cNanotechnology and Integrated Bio-Engineering Centre, School of Engineering, University of Ulster, Newtownabbey, UK BT37 0QB
First published on 8th October 2010
Abstract
Water soluble graphene with various chemical- and biofunctionalities is essential for their different applications. However, exfoliated graphenes are insoluble in water and water soluble graphene oxide precipitate if they are chemically reduced to graphene. We have developed a polyacrylate coating method for graphene oxide and then chemically reduced it into graphene. We found that polyacrylate coating can improve the colloidal stability of both graphene and graphene oxide. The coated graphene has been characterized using XPS, FTIR, XRD and micro-Raman spectroscopy. The primary amine present on the coating backbone has been used to derive glucose functionalized water soluble graphene. Various other functional graphenes can be anticipated from the polyacrylate coated graphene.
Introduction
Graphene has stimulated an enormous research interest in recent years due to its excellent mechanical, electrical, thermal and optical properties and due to very high surface area.1 Graphene based functional materials have significant potential in energy and biomedical research such as photocatalysis, electrochemical detection, drug delivery, coating and optical sensing.2 Soluble and functional graphenes are essential for such applications. However, synthesis of soluble graphene with different functional molecules is very challenging. Presently, large scale soluble graphenes are prepared by exfoliation from graphite using suitable solvent,3 surfactant4 or by chemical reduction of colloidal graphene oxide produced from graphite oxidation via Hummer's method.5 Exfoliated graphenes are soluble in different organic solvents such as N-methyl-pyrolidone,3ortho-dichlorobenzene,6 N,N-dimethylformamide3 or in aqueous surfactant solution.4 Colloidal graphene oxide can be reduced into graphene using hydrazine,5 NaBH4,5p-phenylene diamine;7 however, their solubility decreases upon reduction and they precipitate from solution. Thus a variety of surface functionalization was performed on colloidal graphene oxide so that they are soluble after reduction. These functionalizations include surface passivation with polystyrene sulfonate,8 didodecylammonium bromide,9 DNA,10 aromatic molecules,11 conjugated polyelectrolyte,12 chiotsan based nanocomposite13 or covalent conjugation with aryl diazonium salt,14p-phenylene diamine,7 amine containing small molecule and polymers.15 However, none of these approaches explored the synthesis of water soluble graphene with bioaffinity molecules; either due to the absence of chemical functionality useful for bioconjugation or due to poor colloidal stability of graphene.We are developing various types of coating chemistry for hydrophobic and hydrophilic nanoparticles which are useful in deriving water soluble functional nanoparticles.16 These coating materials include polyacrylate,16f–h imidazole based polymer,16e chitosan oligosaccharide,16c polyaspartate16d and silica.16a,b Among them polyacrylate coating provides the best colloidal stability of coated particle and thus a variety of functional nanoparticle are synthesized using this coating.16f–h,17 Here we have extended this polyacrylate coating to colloidal graphene oxide and found that this coating can significantly improve the colloidal stability of both graphene oxide and graphene. In addition, primary amine present on the coating backbone can be used to prepare graphene based different water soluble functional materials.
Experimental
Materials and reagent
Graphite powder (<20 micron), hydrogen peroxide (30%), hydrazine hydrate (98%), Igepal CO-520, 3-sulfopropyl methacrylate, polyethyleneglycol methacylate (Mn ∼ 360), bis[2-(methacryloyloxy)-ethyl] phosphate and methylenebisacrylamide were purchased from Aldrich and used as received. N-(3-aminopropyl) methacrylamide hydrochloride was purchased from Polyscience.All UV-visible spectra were measured using Shimatzu UV-2550 spectrophotometer with a quartz cell of 1 cm path length. Fourier transform infrared (FTIR) spectra were measured with Nicolet 6700 (Thermo Scientific) instrument using KBr plates. Transmission electron microscopic (TEM) studies were performed with FEI Tecnai G2 F20 microscope and samples were prepared by putting a drop of particle dispersion on a carbon coated copper grid. Diluted graphene oxide (GO) and graphene (G) solution was dropped onto silicon substrate and dried at room temperature to prepare specimens for X-ray photoelectron spectroscopy, Micro-Raman spectroscopy and X-ray diffraction measurements. A Perkin-Elmer PH1-1600 X-ray photoelectron spectrometer (XPS) was used for compositional analysis and chemical bond determination of GO and G. Micro Raman studies were performed using an ISA Lab Raman system equipped with 514.5 nm LASER with a 100× objective giving a spot size of about 1 μm with a spectral resolution better than 2 cm−1. X-Ray diffraction pattern was measured at room temperature using Shimadzu, XRD-6000, 40 kV, 20 mA with Cu K radiation, λ = 1.5418 Å. Diluted GO/G was deposited on glass slide and then AFM was measured using VEECO DICP II autoprobe (model AP 0100). Zeta-potential measurements were performed with a Zetasizer nanosystem (Malvern Instruments Ltd.) using GO/G solutions.
Preparation of graphene oxide
200 mg graphite powder, 100 mg sodium nitrate and ∼5 mL concentrated H2SO4 were mixed and cooled to 0 °C. Then the solution was kept under vigorous stirring. Next, 600 mg KMnO4 was added to this solution in stepwise manner so that the temperature was <20 °C during these KMnO4 addition steps. After the complete addition of KMnO4 the temperature of the solution was slowly raised to 35 °C and kept in this condition for 30 min. A brownish grey paste was formed. Next, ∼10 mL water was added to the whole solution and the solution turned brownish yellow. The temperature of the solution was increased to 98 °C during water addition and this temperature was maintained for 15 min. The whole solution was then mixed with 28 mL of warm water followed by addition of 500 μL 3% H2O2 that reduces the residual permanganate. The light yellow particles were washed thoroughly with warm water 7–8 times. The solid was air dried and dissolved in ∼20 mL distilled water by 15 min sonication. Then it was centrifuged at 3000 rpm for 30 min. The supernatant was collected as graphene oxide (GO) solution and used for polyacrylate coating.Preparation of polymer coated graphene oxide (GO)
Four sets of 2 mL micro-centrifuge tubes were used. In one of them 100 μL of N-(3-aminopropyl) methacrylamide hydrochloride solution (1.8 mg dissolved in 1 mL water) was inserted. In a similar way, 100 μL of polyethyleneglycol methacrylate (360 μL dissolved in 1 mL water), 100 μL of bis[2-(methacryloyloxy)-ethyl] phosphate (50 μL dissolved in 1 mL of H2O) and 100 μL of 3-sulfopropyl methacrylate potassium salt (245 mg dissolved in 1 mL water) were inserted separately in three other micro-centrifuge tubes. Then 0.5 mL of Igepal and 1.4 mL of cyclohexane were added to each micro-centrifuge tube and the solutions were shaken vigorously to make them optically clear. Next, monomeric reverse micelle solutions of the four micro-centrifuge tubes were added to a three-neck flask containing 2 mL of cyclohexane. Then 500 μL of concentrated aqueous graphene oxide solution was added to the reaction mixture and 100 μL of organic base (N,N,N,N-tetramethyl ethylenediamine) was added to the reaction mixture. The whole solution was optically clear and was kept under magnetically stirred conditions and purged with nitrogen for ∼15 min to make the reaction mixture O2 free. After that ammonium persulfate solution (3 mg dissolved in 100 μL of H2O) was injected to the reaction mixture as initiator. The reaction was continued for 1 h in N2 atmosphere and then was quenched by adding a small amount of ethanol. Ethanol addition precipitates the polymer coated graphene oxide which was washed repeatedly with CHCl3 and ethanol. Next they were dissolved in doubly distilled water. In order to remove the unbound polymer, this solution was then centrifuged at 16000 rpm for 10 min and the precipitated particles were redispersed in fresh water. This solution was used for further applications.Preparation of graphene (G) via reduction of graphene oxide (GO)
10 mL of the polymer coated graphene oxide solution was taken in a glass vial and 1 mL hydrazine hydrate (10% of the original volume) was added to it. The solution was then heated under stirring at 70 °C for 30 min. The solution slowly turned black during this time. This solution was kept in room temperature overnight to ensure complete reduction and then dialyzed overnight to remove free hydrazine. The dialyzed solution was then used for further application.Preparation of functional GO and G
Glucose functionalization was performed as reported earlier.16g In brief, polymer coated GO/G solution was taken in carbonate buffer solution and mixed with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of glucosamine and gluteraldehyde solution, which was prepared 15 min earlier. After 1 h borohydride solution was added to reduce the imine bond formed between amine and aldehyde. The solution was then dialyzed to remove free reagents.
:1 molar mixture of glucosamine and gluteraldehyde solution, which was prepared 15 min earlier. After 1 h borohydride solution was added to reduce the imine bond formed between amine and aldehyde. The solution was then dialyzed to remove free reagents.
Results
The polyacrylate coating method and functionalization are shown in Scheme 1. Colloidal graphene oxide was prepared first using Hummer's method.5 Next, it was coated with polyacrylate in reverse micelle medium using our previous approach.16h Typically, concentrated aqueous solution of graphene oxide and acrylate monomers are dissolved in Igepal-cyclohexane reverse micelle to produce an optically clear solution and then polymerization was initiated in oxygen free atmosphere using persulfate initiator. After the polymerization was over, the particles were precipitated using ethanol addition, washed with chloroform and ethanol, and finally dispersed in water. Use of reverse micelle medium has the advantage of solubilizing hydrophilic GO and acryl monomers so that polymerization starts from a homogeneous solution and polymer capping can be very efficient.16h We have tested different acrylate monomers (e.g. N-(3-aminopropyl) methacrylamide, 3-sulfopropyl methacrylate, polyethyleneglycol methacylate, acrylic acid) and different cross linkers (e.g. bis[2-(methacryloyloxy)-ethyl] phosphate and methylenebisacrylamide) and their various combinations. These monomers and cross linkers were selected for testing because of their good water solubility, better capping property and reported better performance in producing water soluble nanoparticle.16f–h Cross-linkers were used in 5–10 mole % in order to minimize the particle–particle linkage during coating. We found that 3-sulfopropyl methacrylate and its combination with polyethyleneglycol methacylate and N-(3-aminopropyl) methacrylamide work well in producing colloidally stable graphene oxide (GO). However, polyethyleneglycol methacrylate, N-(3-aminopropyl) methacrylamide, acrylic acid or their combinations do not produce stable GO solution. Both cross-linkers in 5–10 mol% work well but we used bis[2-(methacryloyloxy)-ethyl] phosphate due to its lower toxicity.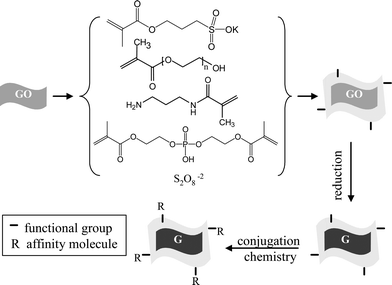 | ||
| Scheme 1 Polyacrylate coating step for colloidal graphene oxide (GO) followed by their conversion into graphene (G) and subsequent functionalization. | ||
Polymer coated GO is water soluble and they can be successfully converted into water soluble colloidal graphene (G) via hydrazine reduction. Fig. 1 shows the UV-visible absorption spectra of aqueous solution of as synthesized GO, polymer coated GO and after subsequent chemical reduction into polymer coated G. While as synthesized GO have negligible absorbance, polymer coated GO shows some increased absorbance in 250–400 nm region due to polymer absorption. However, polymer coated G shows characteristic peaks at 375, 300 and 280 nm, suggesting successful conversion into graphene.9 This spectral change is reflected by a color change from brown to black during the hydrazine reduction of polymer coated GO. AFM study was conducted for as synthesized GO and polymer coated GO/G. It shows that as synthesized GO has a typical thickness of about 0.8–0.9 nm but polymer coated GO/G shows non-uniform thickness of about 10–15 nm. (Fig. 2 and ESI†) This result reveals that as synthesized GO is of a single layer and the obtained GO is of micron size. Polymer coating does not change the average size but increases the thickness of GO/G non-uniformly. From the observed thickness of polymer coated GO/G the coating thickness can be derived between 5–8 nm which agrees well with earlier results of polyacrylate coating on nanoparticles.16f–h,17b This result indicates that the increased thickness of polymer coated GO/G is mainly due to a coating polymer layer, although some multilayer graphene is also possible to occur due to graphene aggregation during coating.
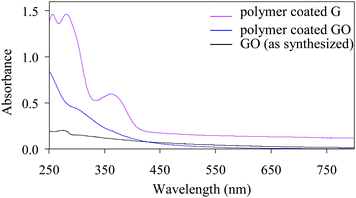 | ||
| Fig. 1 UV-visible absorption spectra of polymer coated graphene oxide (GO) and graphene (G). | ||
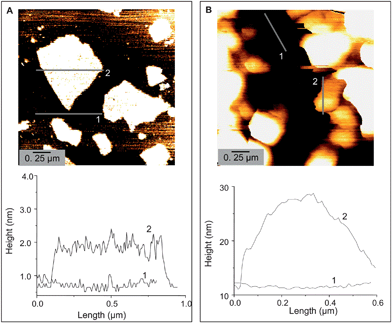 | ||
| Fig. 2 AFM height images of as synthesized GO (A) and polymer coated G (B). | ||
Coating of polyacrylate has been characterized by TGA and FTIR. TGA shows that ∼80 wt% of polymer is bound to GO/G which degrade slowly between 200–400 °C, but the rest of the carbon material remains stable up to 700 °C. (Fig. 3) FTIR study of coated GO/G shows the signature of polymer functional groups. However, the peak intensity at 1200–1500 cm−1 for C–O–C, C–OH functional groups decreases, suggesting the reduction of graphene oxide to graphene.9 (ESI)
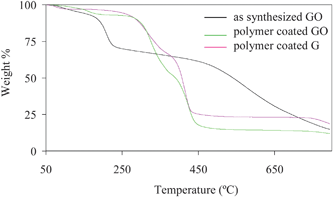 | ||
| Fig. 3 TGA of as synthesized GO, polymer coated GO and polymer coated G. While as synthesized GO suffers 30% weight loss below 200 °C, polymer coated GO and G start losing mass above 300 °C due to polymer degradation, and the weight loss was ∼85% and ∼75%, respectively. This suggests that polymer coating increases the thermal stability of GO and G. Lower weight loss up to 700 °C for polymer coated G as compared to polymer coated GO suggests that G is thermally more stable than GO. | ||
Polymer coated graphene oxide (GO) and their chemical reduction to graphene (G) have also been characterized using XPS, XRD and micro-Raman spectroscopy. As GO and G have polymer coatings, the contribution from the polymer also associates in XPS results and thus having the difficulty in confirming the chemical conversion of GO into G. After carefully analyzing the results and eliminating the signature of polymers we found some indication of transformation (e.g. composition, structure) from GO to G (the details are in the ESI†) XPS based composition analysis shows that graphite powder contains mainly elemental carbon and oxygen having 97.6 at.% and 2.4 at.% respectively, whereas polymer coated GO and G show the presence of a small amount (0.2–2%) of N, S and P in addition to C and O. (ESI).† This result is expected assuming that GO and G have polyacrylate layers which are ∼80 wt% and the polymer contain elemental S, and P coming from the monomers. Micro-Raman study shows the presence of G-band and D-band both in graphene oxide (GO) and graphene (G), but G-bands are broadened and D-bands becomes more intense, as compared to graphite powder. (Fig. 4 and ESI†) This indicates the reduction in size of the in-plane sp2 domains, possibly due to the extensive oxidation and structural imperfections created by the attachment of hydroxyl and epoxide groups on the carbon basal plane. The XRD broad peak at 2θ = 23° for both graphene oxide (GO) and graphene (G) and the corresponding layer to layer distance (d-spacing) of about ≈0.3867 nm are slightly higher than graphite powder and due to structural defects (ESI).†
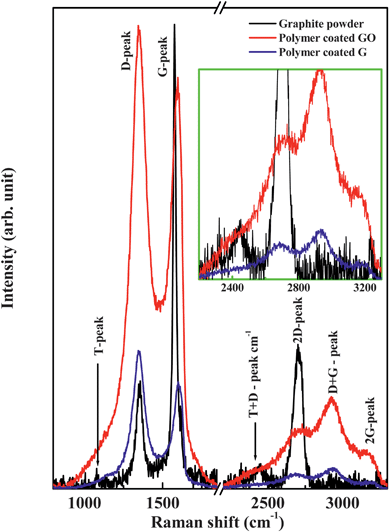 | ||
| Fig. 4 Raman spectra of graphite powder, polymer coated GO and polymer coated G. | ||
Polymer coated water soluble GO and G have good colloidal stability in different aqueous buffer solutions with pH ranging from 5–10 and these solutions are stable for more than a month (Fig. 5). In contrast, as synthesized colloidal graphene oxide has poor solubility in different buffer solutions. In addition, the coated GO and G can be precipitated via high speed centrifuging (typically 16000 rpm with G force of about 25000g) and redispersed in fresh water or buffer solution more than 5 times. These observations suggest that particles are effectively capped and protected from aggregations by polymers. Zeta-potential has been measured for colloidal GO/G in phosphate buffer of pH 7.4, in order to establish their surface charge. While as synthesized GO has a surface charge of −48 mV, it decreases after polymer coating and lies between −10 and −20 mV, depending on the molar ratio of N-(3-aminopropyl) methacrylamide and 3-sulfopropyl methacrylate used during the coating step. For example, when 3-sulfopropyl methacrylate and polyethyleneglycol methacylate were used in 1:1 molar ratio along with 5% cross linker, the surface charge was −18 mV but when 3-sulfopropyl methacrylate, polyethyleneglycol methacylate and N-(3-aminopropyl) methacrylamide were used in 1:1:1 molar ratio the surface charge became −10 mV. The negative surface charge of the as synthesized GO is due to the presence of anionic carboxyl groups formed during the oxidation steps in Hummer's method while the negative surface charge in polymer coated GO/G is due to the presence of anionic sulfopropyl groups. Lowering of the surface charge in polymer coated GO/G, compared to the as synthesized GO, is due to the presence of polyethyleneglycol and primary amine groups on the polymer backbone.
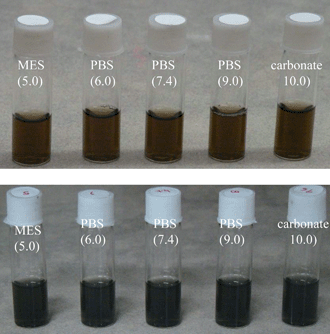 | ||
| Fig. 5 Stable colloidal solution of polymer coated graphene oxide (GO) and graphene (G) in different aqueous buffer solutions. | ||
It is expected that the unique properties of graphene might be modified due to polyacrylate coating. We are particularly interested in the optical sensing based application of colloidal graphene using the strong quenching effect of fluorophore by the graphene surface.2g The polyacrylate coating used here is highly porous in nature17b and thus foreign molecules are expected to be accessible to interact with the graphene surface even after the polyacrylate coating. To test this hypothesis we have studied the fluorescence quenching of rhodamine 6G and the cationic quantum dot (QD) of 20–30 nm size17a by polyacrylate coated graphene (ESI).† We found that polyacrylate coated graphene can efficiently induce fluorescence quenching of rhodamine 6G and cationic QD. However, for QD, quenching is associated with graphene aggregation—due to electrostatic interaction/cross-linking between cationic QD and anionic graphenes. This result shows that polymer coating does not considerably inhibit the access of foreign molecules/particles close to the graphene surface and can be useful for optical sensing based applications.
These colloidal GO and G can be further functionalized with different chemicals and biochemicals via covalent conjugation steps and primary amine groups present on the coating backbone can be used for such purpose. In order to demonstrate this, glucosamine was covalently linked to GO and G via glutaraldehyde based conjugation chemistry and then the biochemical activity of glucose functionalized GO and G was tested via glycoprotein Concanavalein A (Con A) based aggregation assay.16g (Fig. 6) The polyacrylate coating helps to stabilize the GO and G dispersion during the chemical conjugation and purification steps such as dialysis, precipitation via high speed centrifuge followed by redispersion in fresh water. However, the UV-visible absorption spectrum becomes broad after glucose functionalization with a single peak around 290 nm which might be due to surface modification or alteration of π-conjugations of graphenes.9 As Con A has four glucose binding sites, it induces the aggregation of the glucose functionalized soluble graphene. (Fig. 6) The presence of polyethylene glycol in the coating helps minimize the non-specific binging problem during glucose-Con A interaction and thus glucose functionalized graphene does not precipitate in the presence of other proteins such as BSA. This result indicates that other water soluble and functional GO/G can be synthesized using this approach.
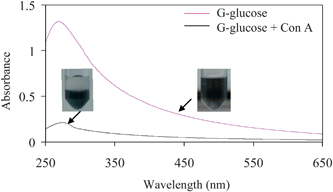 | ||
| Fig. 6 Biochemical activity test of glucose functionalized graphene solution. The dispersed graphenes aggregate and precipitate from the solution after addition of concanavalin A, which induces particle–particle linking via its four glucose binding sites. | ||
Discussion
The goal of this work was to prepare a functional graphene solution of good colloidal stability and to investigate if polyacrylate coating can help in this regard. The motivation comes from the successful synthesis of functional quantum dot, metal and metal oxide nanoparticles using this polyacrylate coating.16f–h,17 Usually the chemical reduction of colloidal graphene oxide leads to precipitated graphene but our polyacrylate coating offers a solution to this problem. When polyacrylate is present as coating, the colloidal graphene oxide does not precipitate upon conversion into colloidal graphene by hydrazine reduction. In addition, coated graphene and graphene oxide possess good colloidal stability during chemical conjugation steps and other adverse physical and chemical conditions. This is because the particles are trapped inside the cross-linked polymer shell. However, successful conversion requires mild heating (60–80 °C) conditions during hydrazine reduction for a short time (30 min) followed by overnight reduction. We observed that the heating at higher temperature for longer time leads to partial precipitation of the particles. This result suggests that there is room for further improvement of this coating. Nevertheless, the advantages of this coating are that it offers one-step solution to modify the surface of graphene by different chemical functionalities (such as primary amine, polyethylene glycol) and variation in their numbers and ratio.16g This can be achieved by using different acrylate monomers or by varying their molar ratio. As the coating method has been used for different types of nanoparticle and acrylate monomers,16f–h coating can be extended to prepare graphene of different surface charges, thermo-responsive graphene or graphene-based composite nanoparticles.We have used the well known Hummer's method for the synthesis of colloidal graphene oxide and the well established hydrazine reduction method for their conversion into graphene.5 It is well known that the outermost basal planes of graphite and edge plane are oxidized during graphene oxide synthesis. The subsequent re-reduction by hydrazine is very efficient in reducing them fully,5 however some structural defects might remain. The re-reduction by hydrazine can be followed by a color change9 and we have also observed that, as shown in Fig. 1. As our coating is porous in nature, small hydrazine molecule can easily access the graphene oxide surface and convert into graphene. The non-uniform thickness of ∼5–8 nm, as observed for polymer coated GO/G, is mainly associated with the coating nature which is also observed for other nanoparticles coated with polyacrylate.16f–h The chances of graphene aggregation during the coating step and the formation of multilayer coated graphene are less, as we have used a standardized coating condition developed earlier.16f–h,17b
The unique properties of graphene have been used in different biomedical research such as photocatalysis, electrochemical detection, drug delivery, coating and optical sensing.2 However, the problem of having highly dispersed graphene and in functional form have limited their application on a wide scale and so far most of the applications used a film/electrode based platform. The presented polyacrylate coating offers attractive alternative options for preparing soluble and functional graphenes that can be used for different solution based applications. Our coating is porous in nature and foreign molecules can easily access the colloidal graphene surface. We have also sown that polymer coated G solution acts as efficient fluorescent quencher. Thus polymer coated G can be useful in optical sensing based specific applications. However, our polyacrylate coating is non-uniform and generally 1–10 nm in thickness, as observed from our earlier studies17b and also from the AFM study of our polymer coated G and GO samples. Thus further improvement of coating might be required prior to application of these coated graphenes in FRET based applications.
Conclusion
We have synthesized functionalized graphene and graphene oxide solution with good colloidal stability. While the conversion of colloidal graphene oxide to graphene by chemical reduction leads to precipitated graphene, polyacrylate coating around graphene oxide can significantly protect this precipitation and provide successful preparation of colloidal graphene. In addition, the polyacrylate coating helps in deriving various water soluble functional graphenes by linking affinity molecules with polyacrylate backbone. The synthesis of water soluble functional graphene opens the door for solution-based utilization of graphene in different biomedical applications.Acknowledgements
This work is financially supported by DST, the Government of India. S.B. and A.S. acknowledge CSIR, India for providing fellowship.References
- (a) S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS; (b) D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS; (c) A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS; (d) C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS; (e) C. N. R. Rao, A. K. Sood, R. Voggu and K. S. Subrahmanyam, J. Phys. Chem. Lett., 2010, 1, 572–580 Search PubMed.
- (a) G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS; (b) J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137–3140 CrossRef CAS; (c) X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 Search PubMed; (d) X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CrossRef CAS; (e) X. Yang, X. Zhang, Y. Ma, Y. Huang, Y. Wang and Y. Chen, J. Mater. Chem., 2009, 19, 2710–2714 RSC; (f) F. M. Koehler, N. A. Luechinger, D. Ziegler, E. K. Athanassiou, R. N. Grass, A. Rossi, C. Hierold, A. Stemmer and W. J. Stark, Angew. Chem., Int. Ed., 2009, 48, 224–227 CrossRef CAS; (g) C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS; (h) Z. Wang, X. Zhou, J. Zhang, F. Boey and H. Zhang, J. Phys. Chem. C, 2009, 113, 14071–14075 CrossRef CAS; (i) J. L. Sabourin, D. M. Dabbs, R. A. Yetter, F. L. Dryer and I. A. Aksay, ACS Nano, 2009, 3, 3945–3954 CrossRef CAS; (j) I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577–583 CrossRef CAS; (k) Q. He, H. G. Sudibya, Z. Yin, S. Wu, H. Li, F. Boey, W. Huang, P. Chen and H. Zhang, ACS Nano, 2010, 4, 3201–3208 CrossRef CAS; (l) X. Wang, L. J. Zhi and K. Mullen, Nano Lett., 2008, 8, 323–327 CrossRef CAS; (m) Z. Yin, S. Wu, X. Zhou, X. Huang, Q. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307–312 CrossRef CAS; (n) B. Li, X. Cao, H. G. Ong, J. W. Cheah, X. Zhou, Z. Yin, H. Li, J. Wang, F. Boey, W. Huang and H. Zhang, Adv. Mater., 2010, 22, 3058–3061 CrossRef CAS; (o) J. Liu, Z. Yin, X. Cao, F. Zhao, A. Lin, L. Xie, Q. Fan, F. Boey, H. Zhang and W. Huang, ACS Nano, 2010, 4, 3987–3992 CrossRef CAS.
- (a) Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS; (b) A. B. Bourlinos, V. Georgakilas, R. Zboril, T. A. Steriotis and A. K. Stubos, Small, 2009, 5, 1841–1845 CrossRef CAS.
- (a) M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS; (b) S. De, P. J. King, M. Lotya, A. O'Neill, E. M. Doherty, Y. Hernandez, G. S. Duesberg and J. N. Coleman, Small, 2010, 6, 458–464 CrossRef CAS.
- (a) S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS; (b) S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS; (c) D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC; (d) W. Chen and L. Yan, Nanoscale, 2010, 2, 559–563 RSC.
- C. E. Hamilton, J. R. Lomeda, Z. Sun, J. M. Tour and A. R. Barron, Nano Lett., 2009, 9, 3460–3462 CrossRef CAS.
- Y. Chen, X. Zhang, P. Yu and Y. Ma, Chem. Commun., 2009, 4527–4529 RSC.
- S. Stankovich, R. D. Piner, X. Q. Chen, N. Q. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- Y. Liang, D. Wu, X. Feng and K. Mullen, Adv. Mater., 2009, 21, 1679–1683 CrossRef CAS.
- A. J. Patil, J. L. Vickery, T. B. Scott and S. Mann, Adv. Mater., 2009, 21, 3159–3164 CrossRef CAS.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Mullen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- X. Qi, K.-Y. Pu, X. Zhou, H. Li, B. Liu, F. Boey, W. Huang and H. Zhang, Small, 2010, 6, 663–669 CrossRef CAS.
- X. Yang, Y. Tu, L. Li, S. Shang and X.-M. Tao, ACS Appl. Mater. Interfaces, 2010, 2, 1707–1713 Search PubMed.
- (a) Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS; (b) J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS.
- (a) S. Park, D. A. Dikin, S. T. Nguyen and R. S. Ruoff, J. Phys. Chem. C, 2009, 113, 15801–15804 CrossRef CAS; (b) C. Shan, H. Yang, D. Han, Q. Zhang, A. Ivaska and L. Niu, Langmuir, 2009, 25, 12030–12033 CrossRef CAS; (c) H. Yang, C. Shan, F. Li, D. Han, Q. Zhang and L. Niu, Chem. Commun., 2009, 3880–3882 RSC; (d) H. Yang, F. Li, C. Shan, D. Han, Q. Zhang, L. Niu and A. Ivaska, J. Mater. Chem., 2009, 19, 4632–4638 RSC.
- (a) S. O. Obare, N. R. Jana and C. J. Murphy, Nano Lett., 2001, 1, 601–603 CrossRef CAS; (b) N. R. Jana, C. Earhart and J. Y. Ying, Chem. Mater., 2007, 19, 5074–5082 CrossRef CAS; (c) E. Nandanan, N. R. Jana and J. Y. Ying, Adv. Mater., 2008, 20, 2068–2073 CrossRef CAS; (d) N. R. Jana, E. Nandanan, J. Jiang and J. Y. Ying, Langmuir, 2010, 26, 6503–6507 CrossRef CAS; (e) N. R. Jana, P. K. Patra, A. Saha, S. Basiruddin and N. Pradhan, J. Phys. Chem. C, 2009, 113, 21484–21492 CrossRef CAS; (f) Y. Wei, N. R. Jana, S. J. Tan and J. Y. Ying, Bioconjugate Chem., 2009, 20, 1752–1758 CrossRef CAS; (g) A. Saha, S. Basiruddin, R. Sarkar, N. Pradhan and N. R. Jana, J. Phys. Chem. C, 2009, 113, 18492–18498 CrossRef CAS; (h) S. Basiruddin, A. Saha, N. Pradhan and N. R. Jana, Langmuir, 2010, 26, 7475–7481 CrossRef CAS.
- (a) S. J. Tan, N. R. Jana, S. Gao, P. K. Patra and J. Y. Ying, Chem. Mater., 2010, 22, 2239–2347 CrossRef CAS; (b) S. Basiruddin, A. Saha, N. Pradhan and N. R. Jana, J. Phys. Chem. C, 2010, 114, 11009–11017 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of XPS, XRD, AFM and FTIR of polymer coated GO and G and results of fluorescence quenching experiments. See DOI: 10.1039/c0nr00376j |
| This journal is © The Royal Society of Chemistry 2010 |