Single-step bifunctional coating for selectively conjugable nanoparticles†
Valerio
Voliani
*ab,
Stefano
Luin
a,
Fernanda
Ricci
ab and
Fabio
Beltram
ab
aNEST, Scuola Normale Superiore and Istituto Nanoscienze-CNR, Piazza San Silvestro, 12, I-56127, Pisa, Italy. E-mail: v.voliani@sns.it
bIIT@NEST, Center for Nanotechnology Innovation, Piazza San Silvestro, 12, I-56127, Pisa, Italy
First published on 11th October 2010
Abstract
A single-step method to coat and bifunctionalize water-reduced gold nanoparticles (NPs) with two distinct reactive groups is reported. The coating is based on a peptide that bonds to the NPs surface by its N-cysteine amino acid, terminates with a C-terminal lysine, and stabilizes the colloids, thanks to the surface organization provided by the rest of the non-polar chain. The process yields stable, non-cytotoxic NPs presenting reactive amine and carboxylic groups on the surface; these allow rapid, selective and modular conjugation of virtually any chosen biomolecule or fluorophore. Functionalized and conjugated nanostructures are analyzed by electrophoresis, SEM, SERS; their biocompatibility and delivery capability are tested by cellular-uptake experiments.
Introduction
Nanomaterials are truly revolutionizing many areas of science. In particular metal nanostructures are emerging as very promising tools to shed light on cellular processes1 through their peculiar localized surface plasmons (LSPs) whose resonance wavelengths are in the near UV-visible range.2,3 Among these, gold nanoparticles (NPs) are increasingly attractive in biomedicine4–6 and biophysics.7,8One of the most important properties of NPs is the possibility to finely regulate their optical and physical behavior directly from their wet chemistry synthesis.9 However, while optical properties are dependent on the nanoparticle composition, shape and size,10,11 the stability and reactivity of the structures depend on the coating of the metallic surface.12 To date many coating methods for water-synthesized NPs were reported that are often lengthy, applicable only to small or specific nanostructures, and/or with a single exposed reactive group or other molecular moiety of interest;13–15 in particular, previous attempts to obtain bifunctionalized coating using peptides terminating with a lysine led to immediate nanoparticle aggregation.16 In order to improve NP versatility, a direct and simple process to achieve bifunctionalized nanostructures with the possibility to “click-like” couple most biomolecules of interest is desirable; by “click-like” we mean the possibility to perform highly efficient, specific, and orthogonal reactions in a range of environments under relatively mild conditions with simple purification procedures.17 The resulting nanoparticles would then be suitable to produce polyvalent nanostructures that can, e.g., incorporate targeting and delivery moieties on the same NP in order to recognize specific cells or cellular compartments, while delivering drugs or being used to accomplish intracellular imaging.14,18
In this article we present a single-step protocol yielding biocompatible water-stable gold nanospheres that exhibit reactive carboxy and amine surface groups. With the present approach we obtain nanostructures that are stable, thanks to the high surface density of the coating, and are readily and modularly conjugable with up to two distinct chosen biomolecules and fluorophores. This method can be applied to other types of nanoparticles such as nanorods, nanoshells and nanocages, and the simple process is suitable for large-scale production of functionalized, versatile nanoparticles. The reaction is carried out on acrylate–acrylic acid reduced nanostructures19 using a short peptide K (CLPFFK), which terminates with a C-lysine and was designed in order to bond to the NPs by the N-cysteine amino acid and to stabilize the coated NPs through the high level of surface organization of the rest of the chain.16,20
To ensure that the amine groups on the peptide are indeed superficial and reactive, the NPs were conjugated to biotin–NHS and analyzed by scanning electron microscopy (SEM) on streptavidinated glass. In this way we demonstrate the success of the process and the stability of the NPs despite the presence of a large amount of amines on the coating. In order to demonstrate the versatility of these NPs, they were conjugated with one or two kinds of fluorophores on one or both of the carboxylic and amine groups. The success of the reaction was tested by surface enhanced Raman scattering (SERS) measurements on the colloids. Finally, biological stability and biocompatibility were tested in vivo by streptavidin-conjugated nanostructures linked to Strep–EGFP. The last system was obtained through EDC/NHS chemistry in a soft reaction environment that does not unfold streptavidin. This shows the performance of the coating here proposed and its usefulness to build new in vivo multitasking biomarkers/sensors within a single nanostructure.
Experimental section
Materials
Hydrogen tetrachloroaurate(III) trihydrate (99.99%) was purchased from Alfa Aesar. Peptides (CLPFFK and CLPFFN, both >95%) were obtained from Caslo Laboratory. Sodium acrylate (97%), acrylic acid (99%), sodium carbonate monohydrate (>99.5%), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, >98%), N-hydroxysuccinimide sodium salt (NHS >98%), biotin–NHS, BSA–biotin, L-glutathione reduced (>98%) and Amicon centrifugal filters were purchased from Sigma-Aldrich. Fluorescein cadaverine, Alexa Fluor® 488 carboxylic acid–succinimidyl ester, and Escherichia coli BL21 (DE3) from Invitrogen. Dialysis membranes from SpectraPore. StrepTactin affinity column, streptavidin and pPR-IBA2 vector from IBA. Mammalian U2OS cell lines from ATCC. All chemicals were used as received without further purification.Synthesis of gold nanospheres
Gold seeds were prepared19 by refluxing for 30 min 100 mL of aqueous solution of HAuCl4 (1 mM) with sodium acrylate (24 mM). The reaction produced a deep-red colloid characteristic of 14 nm spheres. For the second step all the seed solution was mixed to 15 mL of 25 mM HAuCl4 solution and diluted in 845 mL of MilliQ water. After the mixture was brought to pH 7, 40 mL of 0.5 M acrylic acid solution were added, and in 3 days at 22 °C under continuous stirring the reaction produced a wine-red solution of 30 ± 1.7 nm gold nanospheres (AuNs, see Fig. S1†) at a 0.5 nM (2.92 × 1011 NP mL−1) concentration, as observed by ultraviolet-visible (UV-Vis) spectra. The spectra were measured on a Jasco 550 spectrophotometer (Jasco, Tokyo, Japan) supplied with a Jasco ETC-505T thermostat; the colloidal concentration was calculated with the known molar extinction coefficient.10,21Peptide substitution
The native solution of AuNs was rinsed thrice with a 10 mM carbonate solution at pH 9 using Amicon-15 100K centrifugal filters. The bifunctionalized nanospheres (AuNsK) were prepared by mixing 1 mL of a 1 mM stock solution of the CLPFFK peptide (K, Fig. 1) in 1 mM NaCl with 9 mL of the above colloid at room temperature (RT) under stirring for 90 min. The resulting encapsulated nanostructures were purified from non-conjugated molecules by four washing steps in 10 mM carbonate solution at pH 9 with Amicon-15 100K, followed by 1 day of dialysis (SpectraPore membrane 10 K) against carbonate buffer. Assuming that each peptidic chain covers about 0.2 nm2 of AuNs surface,22 the substitution process was carried out in the presence of more than 100-fold excess of peptide with respect to the concentration of nanospheres in order to ensure rapid and thorough superficial coverage.20 The substitution with L-glutathione on AuNs, and the tests on substitution between different peptides (Fig. S6†), were done with similar procedures and concentrations.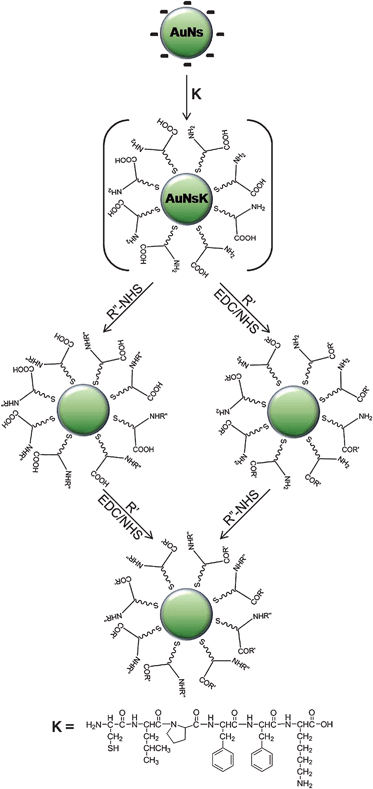 | ||
| Fig. 1 Scheme of the coating process with peptide K (CLPFFK) on water-synthesized gold nanospheres (AuNs) and reactions to conjugate biomolecules and fluorophores onto the new coating (not to scale). Abbreviations: EDC and NHS: see text, R′: fluorescein cadaverine (F) or streptavidin (S), R″-NHS: Alexafluor-488–NHS (A–NHS) or biotin–NHS (B–NHS). | ||
Reactivity of superficial carboxylic groups
To obtain AuNsKF (or AuNsNF), 50 µL of 0.021 M EDC/NHS fresh water solution were mixed to 1 mL of AuNsK (or AuNsN) with 10 µL of fluorescein cadaverine 0.12 M in DMF, continuously mixing at RT for 90 minutes. The resulting solution was washed in a centrifuge with Amicon-4 100K (4000 rpm, 6 minutes each) 3 times with 4 mL of a carbonate/EtOH solution 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3 times with 4 mL of 10 mM carbonate pH 9, and kept in dialysis against carbonate for 12 hours to completely remove the excess of non-conjugated molecules.
:1, 3 times with 4 mL of 10 mM carbonate pH 9, and kept in dialysis against carbonate for 12 hours to completely remove the excess of non-conjugated molecules.
In the process used to bond streptavidin, 500 µL of AuNsK were resuspended in 1 mL PBS buffer pH 7.3 before the reaction, in order to avoid unfolding of the protein. 0.39 mg of lyophilized streptavidin was added to the above colloid with 1 µL of 0.021 M EDC/NHS fresh water solution. After mixing at RT for 180 minutes, the resulting solution was washed in centrifuge with Amicon-4 100K (4000 rpm, 6 minutes each) 4 times with 4 mL of PBS solution at pH 7.3. The functionalized nanostructures were resuspended in 500 µL 10 mM carbonate at pH 8. Control samples were made with the same process but without the presence of EDC/NHS solution.
Reactivity of superficial amine groups
To 500 µL of AuNsK were added 10 µL of 120 mM DMF solution of AlexaFluor-488–NHS or biotin–NHS. After continuously mixing at RT for 90 minutes, the resulting solution was washed in centrifuge with Amicon-4 100K (4000 rpm, 6 minutes each) 3 times with 4 mL of carbonate/EtOH solution 4:1, 3 times with 4 mL of 10 mM carbonate at pH 9, and kept in dialysis against carbonate for 12 hours to completely remove the excess of non-conjugated molecules. Control samples were made with the same process but with non-NHS-modified reactants.
Bifunctionalized NPs
For AuNsKFA, to 500 µL of AuNsKF were added 10 µL of 120 mM DMF solution of AlexaFluor-488–NHS (A). For AuNsKAF, 50 µL of 0.021 M EDC/NHS fresh water solution were added to a mix of 500 µL of AuNsKA and 10 µL 0.12 M of fluorescein cadaverine (F) in DMF. In both cases, after continuous mixing at RT for 90 minutes, the resulting solutions were washed in centrifuge with Amicon-4 100K (4000 rpm, 6 minutes each) 3 times with 4 mL of carbonate/EtOH solution 4:1, 3 times with 4 mL of 10 mM carbonate at pH 9, and kept in dialysis against carbonate for 12 hours in order to exhaustively purify the solutions.
ζ-Potential measurements
Usually 1 mL of the colloidal solutions was centrifuged for 20′ at 13000 rpm and resuspended in the same volume of buffer (carbonate 10 mM pH 9, PBS buffer pH 7.5 or MES 10 mM pH 5.5). The measurements were performed in standard capillary cells DTS 1060 by Zetasizer nano ZS DLS (Malvern Instrument), following manufacturer instructions, in solutions at different pH values (carbonate buffer pH 9, PBS buffer pH 7.5, MES buffer pH 5.5). Each value reported in Fig. S4† is the average of five consecutive measurements.
Scanning electron microscopy
A microscopy glass was incubated at RT for 15 minutes with a 1 mg mL−1 water solution of BSA–biotin. After one washing (with 500 µL of milliQ water) and drying step, a 1 mg mL−1 water solution of streptavidin was stored on the biotinilated surface 15 minutes at RT. Samples of biotin-modified nanospheres (AuNsKB) and controls were stored on the modified surface for different amounts of time (5 to 180 min, Fig. S2†). After a washing step with 500 µL milliQ water and drying with N2, a 5 nm layer of Pd was spread on the surface with a KJL Sigma evaporator. In order to observe the “bare” nanoparticles, we let dry a drop of the colloid on N-doped Silicon (Fig. S1†, bottom). The samples were investigated in a scanning electron microscopy (SEM) Ultra Plus Zeiss.Expression and purification of recombinant green fluorescent protein
In order to obtain fluorescent proteins carrying the biotin mimic peptide MASWSHPQFEKGA (streptag), the EGFP cDNA was subcloned in pPR-IBA2 vector. Recombinant EGFP was expressed in E. coli BL21 (DE3) strain. The maximum yield was obtained harvesting 20 h after induction with isopropyl-β-D-galactoside at 30 °C, as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on the total lysate. Protein was purified by affinity chromatography following manufacturer instructions. A further purification step was carried out by dialysis against PBS buffer to remove the biotin content and to concentrate the protein. The purity was >95%, as judged by SDS-PAGE gels.Cell culture and uptake of nanostructures
U2OS cells were cultured in DMEM medium, supplemented with 10% fetal calf serum at 37 °C, in 5% CO2. Cells were plated two days before imaging onto 30 mm glass-bottom dishes. Nanostructures uptake was achieved by incubating the cells with AuNsKS–StrepGFP system (or with the controls: AuNsK–StrepGFP or StrepGFP) for 1 hour at 37 °C, in 5% CO2. U2OS cells were imaged both after having been washed three times with PBS buffer and rinsed with fresh complete medium either not. Cell viability was tested using tryptan blue assay coupled with automatic cell counting system (countesstm—Invitrogen). Cell-membrane-integrity assay was performed on washed samples with 5 µg mL−1 of Propidium Iodide (PI) for 20 min at 37 °C, in 5% CO2.Image acquisition
Confocal images were acquired with a Leica TCS SP2 on DM-IRE2 inverted confocal microscope (LeicaMicrosystems), using a 40× 1.25 NA oil immersion objective and the 488 nm and 561 nm laser lines for the excitation of EGFP and PI fluorescence, respectively. ImageJ software was used to compute image analysis and background was subtracted from all images.Results and discussion
Peptide coating
In this work we propose an efficient and straightforward method yielding bifunctionalized water-soluble gold nanospheres readily conjugable with a broad spectrum of biomolecules and fluorophores by simple EDC/NHS chemistry. In particular, this method was applied to monodisperse spherical gold nanostructures (average diameter of 30 nm), synthesized using the seeding growth method described by Zhong et al.19Fig. 1 and its caption illustrate schematically the coating process and the abbreviations for the reactants used in this work (e.g. B is biotin); their position in the name of the samples indicates the reaction sequence, e.g. AuNsKFA represents AuNs nanospheres coated with the K peptide and conjugated first with Fluorescein cadaverine (F) and then with AlexaFluor-488 (A).
Encapsulation of the nanostructures is based on a six-amino acid peptide (CLPFFK, K-peptide) comprising three sections: an N-cysteine, a C-terminal lysine and a four non-polar-amino acids sequence. The peptide bonds to the AuNs surface by the N-cysteine amino acid, where the thiol group provides a strong affinity for gold (∼45 kcal mol−1).20 In turn the C-terminal lysine supplies free carboxylic and amine groups on the exposed surface of the nanostructures (Fig. 1), while the rest of the non-polar side chains of the amino acid sequence was designed in order to yield a dense self-assembled monolayer (SAM) that excludes water from the core and therefore stabilizes the system;20 this non-polar SAM changes the average dielectric function around the nanosphere and is therefore responsible for a small shift in the plasmon resonance10 (see Fig. S3†). A reference system was realized replacing the K-peptide with the CLPFFD–NH2 peptide (N in the following), in order to obtain modified NPs with only carboxylic groups on the external surface of the coating. The two peptides have the same amino acidic sequence except for the last amino acid (the N-peptide has an amidated aspartic acid instead of lysine). The success of the substitution process was tested by dynamic light scattering experiments, in particular of the ζ-potential (Fig. S4†), and by measuring the electrophoretic motility in 1% agarose gel at 70 V of AuNs, AuNsK, and AuNsN (Fig. 2, lanes 1, 2 and 3, respectively).
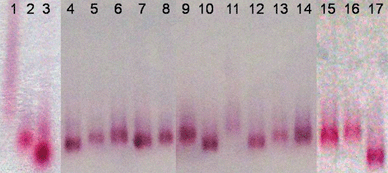 | ||
| Fig. 2 Collage of 1% agarose gel electrophoresis of samples performed for 180 minutes at 70 V. To test the repeatability of the experiment and the homogeneity of the electrical field the sample AuNsK was inserted every 3–5 lanes to use its band as internal reference. Lanes 1: AuNs; 2: AuNsK; 3: AuNsN; 4: AuNsKFA; 5: AuNsKFA control; 6: AuNsK; 7: AuNsKF; 8: AuNsKF control; 9: AuNsK; 10: AuNsKB; 11: AuNsKB control; 12: AuNsKA; 13: AuNsKA control; 14: AuNsK; 15: AuNsK; 16: AuNsKS; 17: AuNsN. See text for the abbreviations. | ||
ζ-Potential at physiological pH goes from −30.4 V for AuNs to −17.8 V for AuNsN and −14.1 V for AuNsK. We measured a dependence of the ζ-potential on pH for AuNsN, and AuNsK, typical of carboxyl only, and of carboxyl and amine groups, respectively (Fig. S4†). Indeed ζ-potential is linked to the total charge on the nanostructures, which depends on pH. While in AuNsN the only protonable sites are the carboxylic groups (reported pKa 2.9), in AuNsK there are also the amino groups (pKa 10.7), which concur to neutralize the total charge on the encapsulated bifunctionalized-nanoparticles.
The success of the substitution process can be implied also from the differences in the electrophoretic mobility shown in the first 3 lanes of Fig. 2. The fact that “bare” AuNs run in a smear and slower than the coated nanoparticles, although they have an higher negative charge (as from the ζ-potential), it's a sign of aggregation.23 The other two samples run in bands with different retentions reflecting the difference in their superficial charge. Electrophoresis was performed in TBE 0.5× (pH 7.5) and, significantly, the bifunctionalized sample shows lower mobility because the partial protonation of the superficial amine reduces the total (negative) charge of the system.
We obtained stable water-soluble nanostructures with both amino and carboxylic surface groups that allow the selective conjugation of other molecules. In order to test the reactivity of these groups on the metallic surface, nanostructures were conjugated by amide bonds to fluorophores and proteins by EDC/NHS chemistry (EDC = N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide, NHS = N-hydroxysuccinimide).24 “Control” samples were also obtained using similar reactions, but without the reactive molecules (i.e. EDC/NHS or NHS-modified molecules); this allowed us to rule out any non-specific adsorption on the metal surface or within the coating. To preliminarily confirm the success of the reactions, 1% agarose gel electrophoretic analysis was performed. Fig. 2 shows that every control sample (lanes 5, 8, 11 and 13) runs like the reference AuNsK bands (lanes 6, 9, 14 and 15), thus demonstrating that no appreciable modification of (or adsorption on) the surfaces occurred. Modified-AuNsK systems show different behaviors because of two major effects, i.e. the variation of hydrodynamic radius and of total charge. The former originates from the volume increase due to the conjugation of the colloids with the selected molecules. Streptavidin modified-AuNsK (AuNsKS) were particularly affected by this phenomenon, because of the large dimension of the protein (57 kDa). In fact Fig. 2 lane 16 shows that AuNsKS is the only sample running slower than the AuNsK bands. Concerning charge variation, the total charge of the bifunctionalized nanostructures (negatively charged at pH > 4) depends on the protonation state of the superficial groups or conjugates. At pH 7.5 an appreciable fraction of superficial amines is positively charged. When negatively (or almost neutrally) charged molecules are conjugated to the amino groups by an amide bond, the contribution of the amine to the total charge of the system is lost; therefore negative charge on the nanostructures increases and enhances the motility in electrophoresis (Fig. 2, lanes 10 and 12). The conjugation on the carboxylic groups with (more) negative fluorophores also increases slightly the total negative charge of AuNsK (Fig. 2, lane 7), and the modification of both the NH2 and COOH groups on the metallic surface produces a synergic effect by increasing further the negative charge (Fig. 2, lane 4).
Immobilization on glass surface was used to confirm the electrophoresis results on modified AuNsK and the prompt reactivity of the superficial amines. To this end, glass surfaces were functionalized with a water solution of BSA–biotin3,25 and streptavidin. Samples of biotin–nanospheres (AuNsKB) and non-conjugated nanostructures (AuNsKB control) with the same concentration were stored on these surfaces for 40 minutes (Fig. 3a). These samples (Fig. 3b) were imaged with a SEM. We observed mostly single particles, demonstrating that no aggregation of the coated and biotinilated nanostructures occurs. We measured the superficial density of AuNsKB by automatically counting the number of nanospheres on randomly selected areas (using ImageJ software). The density of the nanoparticles on the samples was 500 times greater than on controls (respectively, 20.10 NP per µm2 and 0.04 NP per µm2). Significantly we observed a time-dependent density of the AuNsKB deposition (see Fig. S2†). In particular the number of modified nanospheres on the sample increases with deposition time, reaches a maximum after about 40 minutes, and, at greater times, nanoparticles aggregate on the functionalized glass surface and lift off. This behavior suggests the possibility of tuning the AuNs density on the functionalized surfaces.
 | ||
| Fig. 3 (a) Schematic picture of the surface binding between biotin–nanospheres (AuNsKB) and streptavidinated glass (not to scale). First, streptavidin is immobilized on BSA–biotin functionalized glass surface. Then, biotin–gold nanospheres (AuNsKB) are tethered to the immobilized streptavidin; samples without biotin conjugation do not bind to the surface. Finally a 5 nm Pd layer is deposited on the samples for visualization in a scanning electron microscope (SEM). (b) SEM image of AuNsKB (top) and AuNsKB control (bottom) samples on the streptavidinated glass. Nanostructure densities are 20.10 Np per µm2 and 0.04 Np per µm2 after 40 minutes of incubation, respectively (see ESI† for data at different incubation times). | ||
Surface enhanced Raman scattering (SERS)
SERS spectroscopy is a surface-sensitive technique that results in the enhancement of Raman scattering by molecules in proximity of (adsorbed on or linked to) metal surfaces.26 This enhancement is particularly strong when the metallic surfaces possess nanometric-size roughness or features, and it can reach values up to 1014 when appropriate NPs are used,7 while it decreases very rapidly when the distance between the molecule and the surface increases. SERS techniques were recently proposed as suitable for biodetection and diagnosis, given the possibility to attach “barcodes” or recognition elements to NPs.27 Here we shall discuss our results on colloidal solutions where different fluorophores were bonded to the nanospheres through the superficial amines (AuNsKA), or the superficial carboxylic groups (AuNsKF), or both of them (AuNsKFA, AuNsKAF). In this way the reactivity of the different groups introduced on the nanostructures by the bifunctionalized coating can be tested, since only signals from the molecules covalently linked to the nanospheres can be collected. SERS spectra (Fig. 4) were obtained using a setup described elsewhere28 with a Laser 561 Oxyus PS-003 at 100 mW, filters Chroma Z561/10X and Semrock LP02-568RU (laser clean up and long-wave-pass Raman filter, respectively). Spectra were recorded typically with a colloidal concentration of 0.3 nM at pH 8.6 and 30 min integration time.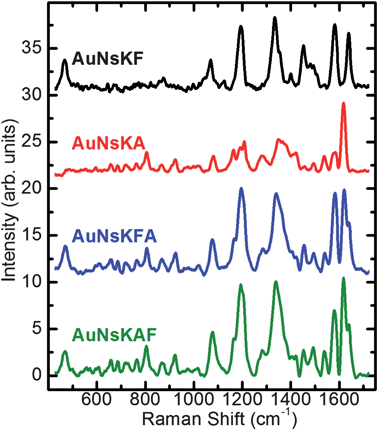 | ||
| Fig. 4 SERS spectra, after background subtraction, collected from colloidal solutions of gold nanospheres conjugated with one or both fluorophores at room temperature (see text for abbreviations and further details). | ||
The black and red lines in Fig. 4 show SERS spectra of AuNsK conjugated with fluorescein cadaverine and Alexafluor-488 (AuNsKF and AuNsKA, respectively). Spectra show specific peaks characteristic of the fluorophores, consistent with those previously reported,29 demonstrating the success of the reactions. The confirmation of the ready-to-use bifunctionalized coating was shown by the conjugation of the gold nanospheres with both Alexafluor-488 and fluorescein cadaverine on the same nanoparticles. Blue and green lines (AuNsKFA and AuNsKAF, respectively) in Fig. 4 show very similar shape and the spectra are virtually the sum of the SERS spectra of the single fluorophores. This result suggests that the order of the reaction process is not important, and that the chemical–physical behavior of the system and of the reactive superficial groups is unaffected by partial conjugation.
Contrary to fluorophore-linked nanospheres, spectra of the controls (nor of bare AuNs) do not exhibit any relevant observable fluorophore-specific structures (data not shown), confirming the absence of non-specific adsorption on the metallic surface; this remains true even when small aliquots of the fluorophore(s) are present in the control solutions, and their fluorescence signal is at similar or even higher level than the one in the fully labeled samples.
Nanoparticles as carriers into living cells
Transfer of particles into cells can be performed in various ways, among them fluid-phase uptake from the culture medium or mechanical methods (e.g., microinjection). Nanostructure cell penetration is regulated by many factors such as nanoparticle shape, size and charge, and its conjugation with biomolecules.30 It was found that negatively charged metal nanostructures can enter cells by endocytosis30,31 with a strong NP-size dependence.32 In a study by Chithrani et al., the maximum efficiency of cellular uptake, without cell-membrane damage, was found for nanoparticles with 30–50 nm diameter.32To verify that the AuNsK system can be useful for the delivery of biomolecules, nanostructures were conjugated to a protein cargo. To this purpose the AuNsKS system was conjugated with a recombinant Green Fluorescent Protein (EGFP, S65T F64L) exploiting a strept-tag (MASWSHPQFEKGA) inserted into its N-terminal. The binding reaction was carried out in PBS buffer in the presence of 25 pM AuNsKS and 100 nM StrepGFP in a final volume of 255 µL. The reaction was allowed to proceed for 60 min at room temperature under continuous stirring. No other purification was employed and the resulting AuNsKS–StrepGFP system was directly used in the uptake experiment. Human liver cells (U2OS) were incubated with AuNsKS–StrepGFP for 1 h at 37 °C, washed three times with PBS, and finally rinsed with fresh medium. Cells were allowed to equilibrate for 10 min; confocal fluorescence images under excitation at 488 nm are shown in Fig. 5. The chromophore of the GFP is rather far from the metallic surface, thanks to the presence of the K-peptide, of the Streptavidin and of the GFP β-barrel; therefore, we do not expect to observe the fluorescence-quenching effect often seen for organic molecules adsorbed on metallic surfaces. Indeed intense GFP fluorescence (495–560 nm) from endosomes in the cytoplasm was observed when the AuNsKS–StrepGFP complex was added to the culture (Fig. 5A, D and E). No fluorescence into cells was observed in the control experiments performed either with AuNsK–StrepGFP (non-conjugated system) or with StrepGFP alone in the same conditions (Fig. 5B and C). These results demonstrate that the conjugation of the StrepGFP to the streptavidin-modified nanoparticles was successfully achieved and necessary for AuNsKS to successfully translocate a cargo (i.e. GFP) into cells. The cell viability test revealed the non-toxicity of AuNsK; moreover, no damage to cell membranes following AuNsKS–StrepGFP uptake was observed by propidium iodide (PI) assays, since no PI staining was observed, in agreement with other studies32,33 (Fig. S5†). We also performed in vitro experiments on the possibility of peptide-release caused from physiological concentration of glutathione, one of the most abundant thiols inside living cells, showing no competition with our system (Fig. S6†). These results indicate that the present functionalized nanoparticles can be of interest as targeting and drug-delivery systems in living cells. The tunability of nanoparticle surface charge and the possibility to incorporate a targeting functionality will allow to further enhance the final efficiency of uptake and intracellular cargo localization.
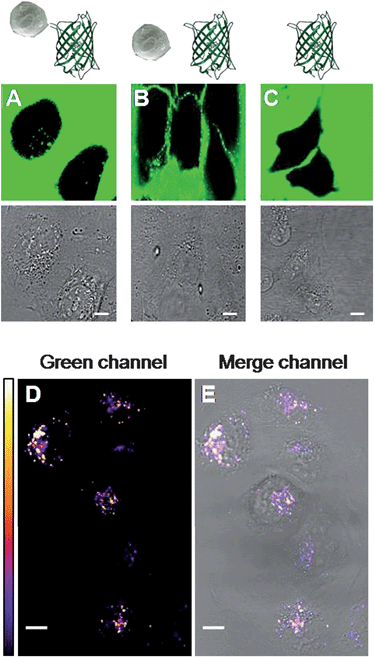 | ||
| Fig. 5 (A–C) Images of human U2Os cells after 1 h of incubation in a medium containing AuNsKS–StrepGFP, AuNsK with StrepGFP, and StrepGFP, respectively (25 pM in nanoparticles when applicable; see the schematic representation in the row above, and text for further details). Top: confocal green channel (excitation at 488 nm, emission at 495–560 nm) showing GFP emission fluorescence; bottom: transmission bright field channel. In the green channel images, the presence of GFP outside the cells is demonstrated by the diffused fluorescence; in A, the uptake of the AuNsKS–StrepGFP is confirmed by the fluorescence from internal organelles, while no internalization of the negative controls (B and C) was observed for these systems after 1 h of incubation. (D) Confocal fluorescence image showing GFP emission fluorescence from the internalized complex after washing. White color indicates higher fluorescence intensity, as shown in the LUT reported on the left. (E) The bright field image merged with the green channel highlights the cytoplasmic localization of the AuNsKS–StrepGFP. For all images scale bar: 10 µm. | ||
Conclusions
In conclusion, we have presented a one-step strategy to encapsulate gold nanospheres with a layer of short C-terminal lysine peptides, allowing their use as quick modular systems that can be rapidly coupled to two of virtually any biomolecule or fluorophore of interest. By this one-step process the metallic nanostructures are stabilized in water, most probably thanks to the formation of a hydrophobic SAM around the surface, and bifunctionalized by amine and carboxylic reactive groups. The success of the coating and the reactivity of the superficial groups were tested and confirmed by electrophoresis, immobilization followed by scanning electron microscopy, and surface enhanced Raman scattering on nanoparticles conjugated with one or two biomolecule(s) or fluorophore(s). We also demonstrated the biocompatibility of the nanostructures linked to proteins by in vivo uptake and cell-viability experiments, where our protocol was tested with the final aim of achieving targeting and drug delivery systems (even at pico-molar concentrations). We underline the versatility of this technique and its straightforward applicability for the production of nanostructures of interest in biomedicine, e.g. as a building block for complex sensing or pharmacologically active nanostructures, or in biophysics, e.g. as targetable probes in a number of configurations taking advantage of fluorescence labelling or light scattering from the NPs themselves.1,18,25Acknowledgements
The authors thank Victor F. Puntes, Liberato Manna, Giovanni Signore, Pasqualantonio Pingue and Paolo Faraci for stimulating discussions. This work was partially funded by the Italian Ministry of Research (MiUR) under project RBLA03ER38 and by Fondazione Monte dei Paschi di Siena.References
- J. Aaron, K. Travis, N. Harrison and K. Sokolov, Nano Lett., 2009, 9, 3612–3618 CrossRef CAS.
- J. Bingham, K. Willets, N. Shah, D. Andrews and R. Van Duyne, J. Phys. Chem. C, 2009, 113, 16839–16842 CrossRef CAS.
- C. Sonnichsen, B. M. Reinhard, J. Liphardt and A. P. Alivisatos, Nat. Biotechnol., 2005, 23, 741–745 CrossRef.
- A. P. Alivisatos, Sci. Am., 2001, 285, 66–73 CrossRef CAS.
- A. Neely, C. Perry, B. Varisli, A. K. Singh, T. Arbneshi, D. Senapati, J. R. Kalluri and P. C. Ray, ACS Nano, 2009, 3, 2834–2840 CrossRef CAS.
- W. Eck, G. Craig, A. Sigdel, G. Ritter, J. Lloyd and M. Mason, ACS Nano, 2008, 2, 2263–2272 CrossRef CAS.
- J. Kneipp, H. Kneipp, M. McLaughlin, D. Brown and K. Kneipp, Nano Lett., 2006, 6, 2225–2231 CrossRef CAS.
- P. S. Ghosh, C. K. Kim, G. Han, N. S. Forbes and V. M. Rotello, ACS Nano, 2008, 2, 2213–2218 CrossRef CAS.
- C. Orendorff, T. Sau and C. Murphy, Small, 2006, 5, 636–639 CrossRef.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- M. Hu, J. Chem, Z. Li, L. Au, G. Hartland, X. Li, M. Marquez and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084 RSC.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS.
- K. Aslan and V. Prez-Luna, Langmuir, 2002, 18, 6059–6065 CrossRef CAS.
- S. Kumar, J. Aaron and K. Sokolov, Nat. Protoc., 2008, 3, 314–320 Search PubMed.
- C. Stroh, H. Wang, R. Bash, B. Ashcroft, J. Nelson, H. Gruber, D. Lohr, S. M. Lindsay and P. Hinterdorfer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12503–12507 CrossRef CAS.
- R. Levy, N. T. Thanh, R. C. Doty, I. Hussain, R. J. Nichols, D. J. Schiffrin, M. Brust and D. G. Fernig, J. Am. Chem. Soc., 2004, 126, 10076–10084 CrossRef CAS.
- B. Sumerlin and A. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- S. Kumar, N. Harrison, R. Richards-Kortum and K. Sokolov, Nano Lett., 2007, 7, 1338–1343 CrossRef CAS.
- P. N. Njoki, I.-I. S. Lim, D. Mott, H.-Y. Park, B. Khan, S. Mishra, R. Sujakumar, J. Luo and C.-J. Zhong, J. Phys. Chem. C, 2007, 111, 14664–14669 CrossRef CAS.
- N. Bastùs, E. Sanchez-Tillò, S. Pujals, C. Farrera, C. Lòpez and V. Puntes, ACS Nano, 2009, 3, 1335–1344 CrossRef CAS.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef CAS.
- T. Zhu, K. Vasilev, M. Kreiter, S. Mittler and W. Knoll, Langmuir, 2003, 19, 9518–9525 CrossRef CAS.
- Y. Chen, H. Liu, T. Ye, J. Kim and C. Mao, J. Am. Chem. Soc., 2007, 129, 8696–8697 CrossRef CAS.
- R. Sperling, T. Pellegrino, J. Li, W. Chang and W. Parak, Adv. Funct. Mater., 2006, 16, 943–948 CrossRef CAS.
- B. M. Reinhard, M. Siu, H. Agarwal, A. P. Alivisatos and J. Liphardt, Nano Lett., 2005, 5, 2246–2252 CrossRef CAS.
- C. E. Talley, J. B. Jackson, C. Oubre, N. K. Grady, C. W. Hollars, S. M. Lane, T. R. Huser, P. Nordlander and N. J. Halas, Nano Lett., 2005, 5, 1569–1574 CrossRef CAS.
- R. A. Alvarez-Puebla and L. M. Liz-Marzan, Small, 2010, 6, 604–610 CrossRef CAS.
- S. Luin, V. Voliani, G. Lanza, R. Bizzarri, P. Amat, V. Tozzini, M. Serresi and F. Beltram, J. Am. Chem. Soc., 2009, 131, 96–103 CrossRef CAS.
- P. D. Lacharmoise, E. C. Le Ru and P. G. Etchegoin, ACS Nano, 2009, 3, 66–72 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 1, 12–21 CrossRef.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM pictures, UV-Vis spectra and DLS measurements of bare, coated and functionalized gold nanospheres, confocal fluorescence images of cell-internalized systems with PI-staining, electrophoresis of AuNs coated with glutathione and of AuNsK and AuNsN in the presence of glutathione. See DOI: 10.1039/c0nr00350f |
| This journal is © The Royal Society of Chemistry 2010 |