Synthesis of porous CuO–CeO2 nanospheres with an enhanced low-temperature CO oxidation activity
Jinwen
Qin
,
Junfeng
Lu
,
Minhua
Cao
* and
Changwen
Hu
*
Key Laboratory of Cluster Science, Ministry of Education of China, Department of Chemistry, Beijing Institute of Technology, Beijing, 100081, P. R. China. E-mail: caomh@bit.edu.cn; cwhu@bit.edu.cn; Fax: +86 (10) 6891263; Tel: +86 (10) 6891263
First published on 13th October 2010
Abstract
CuO–CeO2 nanospheres with a porous structure were synthesized by an improved urea method involving first hydrothermal treatment to get Ce–Cu binary precursor and then the calcination of the precursor. The CuO–CeO2 nanospheres consist of spherical particles with diameters in the range of 300–400 nm. These nanospheres are actually composed of nanoparticles of ca. 10 nm, resulting in the formation of a mesoporous structure. Compared with conventional urea method, in which Ce–Cu binary precursor is commonly achieved in an oil bath at appropriate temperature, the Ce–Cu binary precursor obtained via the hydrothermal process could be more highly homogeneous and more highly interdispersed CuO–CeO2 thus was formed. In addition, the resulted porous CuO–CeO2 catalyst has a lower CO oxidation temperature of as low as 71 °C
1 Introduction
Recently, CeO2 and CeO2-based materials have attracted much attention with respect to their application in catalysis due to their oxygen vacancy defects and high oxygen storage capacity (SOC) resulting from the facile Ce4+/Ce3+ redox cycle. Various CeO2-based binary oxides with the incorporation of varied metal oxides into CeO2 fluorite lattice have proven to exhibit higher SOC and better catalytic performance than pure CeO2.1 Among above mentioned materials, CuO–CeO2 is particularly attractive because of its high catalytic activity toward CO oxidation, which has potential applications in air cleaning and reducing automotive emissions.2 In general, the activity of a catalyst closely depends on its preparation method, structure, and morphology previously reported in the literature, in which porous structure and high specific surface area is particularly effective to enhance its activity.1f As a consequence, a variety of methodologies have been developed to obtain CeO–CeO2 with high catalytic activity via the modification of size, shape, interface, and porosity, such as the template method,3,4 inert gas condensation,5 solution combustion method,6 single-step flame spray pyrolysis,7etc. However, these methods for the synthesis of CuO–CeO2 compounds generally suffer from some harsh conditions, such as the usage of surfactants, templates, organic solvents, hyperthermal treatment and so on. Therefore, finding a facile way to construct this kind of structure remains a big challenge.Herein, CuO–CeO2 nanospheres with a porous structure were synthesized by an improved urea method involving first hydrothermal treatment to get Ce–Cu binary precursor and then the calcination of the precursor. Compared with conventional urea methods, in which the Ce–Cu binary precursor is commonly achieved in an oil bath at an appropriate temperature, the Ce–Cu binary precursor obtained via the hydrothermal process could be more highly homogeneous and thus a more highly interdispersed CuO–CeO2 was formed.8 Furthermore, it is more important that the resulting CuO–CeO2 nanospheres, obtained without the aid of any surfactants, exhibit a mesoporous structure, while recently reported porous binary metal oxides, such as TiO2–SiO2 and TiO–ZrO2, are usually prepared in the presence of surfactants.9 Obviously, the advantage of this method for the synthesis of CuO–CeO2 mesoporous structures is simplicity and low cost, which are very important for large-scale production. So far, the lowest CO oxidation temperature of CuO–CeO2 catalyst reported previously is also as high as 100 °C. For example, Amal's group reported flame-synthesized ceria-supported copper dimers for preferential oxidation of CO, in which complete oxidation achieved at 100 °C.7 In our study, the resulting porous CuO–CeO2 catalyst has a lower CO oxidation temperature of as low as 71 °C.
2 Experimental section
Preparation of CuO–CeO2 nanospheres
CuO–CeO2 nanospheres were prepared by a hydrothermal treatment based on the two-step route. Ce(NO3)3·6H2O (1 mmol) and Cu(NO3)2·3H2O (1 mmol) was dissolved in 50 mL of distilled water, followed by the addition of urea (8 mmol). The resulting solution was stirred to attain a transparent solution, transferred to a Teflon autoclave, and then heated at 180 °C for 100 min, which led to precipitation of the precursor. The precipitate was centrifuged, washed twice with deionized water and once with ethanol. After drying at ambient atmosphere, the precursor finally calcined under air at 600 °C for 4 h. Samples were pale green to pale yellow in color.Characterization
The crystal structure of as-prepared CuO–CeO2 nanocomposites was determined by a Shimadzu XRD-6000 diffractometer with Cu-Kα radiation. SEM image was recorded on a Hitachi X650 microscope. TEM images were obtained by a Philips Tecanai G2F20 system operating at 200 kV. Its surface area and pore size distribution by nitrogen adsorption/desorption at 77 K using Belsorp-max. XPS analysis was performed on a VG Escalabmkll spectrometer (UK). ICP measurements were recorded on a JY Ultima (French).Catalytic tests
The CO oxidation of CuO–CeO2 catalysts heated in situ for activity was carried out with 50 mg in a glass tubular reactor under an atmosphere pressure flow of 30 L h−1 g−1, with a 1% CO and fresh air balanced composition from air generator. The reactor outlet stream was monitored for CO and CO2 concentration by GC 9800. H2-TPR experiments were performed with a thermal conductivity detector on 50 mg samples in 80% (molar) argon and 20% (molar) hydrogen gas mixture, using a gas flow rate of 100 mL min−1 with a temperature ramp rate of 10 K min−1.3 Results and discussion
After calcining the Ce–Cu binary precursor obtained directly by hydrothermal process, CuO–CeO2 composites were obtained. The composites have a Cu/(Cu + Ce) ratio of 33.44% (hereinafter, referred to as CeCu0.33), which has been confirmed by inductively coupled plasma spectroscopy (ICP). Fig. 1a shows the X-ray diffraction (XRD) pattern of the CuO–CeO2 composites. It can be clearly seen from Fig. 1a that all diffraction peaks can be assigned to the cubic phase of CeO2 with lattice constant a = 5.411 Å (JCPDS No. 04-0593) coexisting with the monoclinic phase of CuO with lattice constants a = 4.685 Å, b = 3.423 Å, and c = 5.132 Å (JCPDS No. 41-0254). The two typical reflections centered at 35.62° and 38.82° are usually characteristic for the present of Tenoirte (CuO) phase. In addition, it is highly possible that a slight amount of CuO has been incorporated into the CeO2 lattice due to the high calcination temperature (600 °C).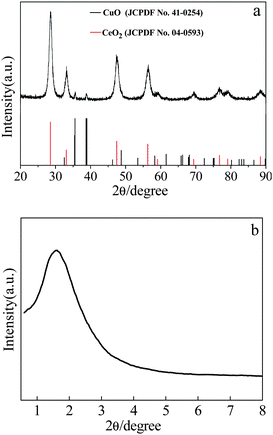 | ||
| Fig. 1 XRD pattern of as-prepared CeCu0.33 nanospheres. | ||
The morphology and microstructures of the prepared CeCu0.33 nanocomposites were investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The low magnification SEM image in Fig. 2a shows that the sample consists of spherical particles with diameters in the range of 300–400 nm. These nanospheres are actually composed of nanoparticles, which are observed from broken nanospheres, as shown in high magnification SEM (Fig. 2b). The TEM image of a single nanosphere is shown in Fig. 3a, which further confirms above deduction. These small nanoparticles consisting of the nanospheres have a diameter of ca. 10 nm. The crystallite sizes prepared by the improved urea method are clearly smaller than those prepared by the conventional urea method, confirmed by the XRD (Fig. 1a) and TEM (Fig. 3b) results. It may be due to the fact that with the modified method the precursor obtained under hydrothermal conditions has a more uniform distribution of Cu and Ce species. The Cu species of uniform distribution in precursor could prevent resulting CeO2 from sintering.
 | ||
| Fig. 2 SEM images of as-prepared CeCu0.33 nanospheres (a) low magnification, (b) high magnification. | ||
 | ||
| Fig. 3 (a) TEM image; (b) high-magnification TEM of as-prepared CeCu0.33 nanospheres; (c) and (d) HRTEM images of as-prepared CeCu0.33 nanospheres in the interior regions showing CeO2 and CuO nanoparticles, respectively, and insets show respective FFT patterns. | ||
Furthermore, the local magnification of the nanosphere, as shown in Fig. 3b, clearly discloses that the nanospheres possess a porous structure. High-resolution TEM (HRTEM) images of different surface locations of the nanosphere give spacings of 0.156 and 0.233 nm, which match well with (222) planes of cubic CeO2 phase and (111) planes of monolinc CuO phase, respectively.10 And respective Fast Fourier Transformation (FFT) patterns (insets in Fig. 3c and 3d) further confirm the above result. Therefore, from the HRTEM images, it can be seen that the nanospheres are mainly composed of mixed crystallites, preferentially exposing the disordered crystal phases of CuO and CeO2 crystallites. Their catalytic effect may benefit by much more oxygen vacancies from the lattice defects and strains deriving from these disordered crystal phases.11
Brunauer–Emmett–Teller (BET) analyses are performed to further investigate the porous structure and surface area of the as-obtained CeCu0.33 nanospheres. N2 adsorption/desorption isotherms and corresponding Barrett–Joyner–Halenda (BJH) pore size distribution plots are shown in Fig. 4. The isotherms can be assigned as type IV with an evident hysteresis loop in the range of 0.45–0.8. The hysteresis profile indicates the typical mesoporous character of this sample (Fig. 4a). The specific surface area estimated from the BET method is 61 m2 g−1, which is evidently superior to that of pure nanoparticles. This fact may result from their highly porous structure. The size of the pores based on desorption data exhibits a wide distribution centered at 1.55, 1.83, 2.4, and 2.2 nm (Fig. 4b), covering the range of both micropores and mesopores. Furthermore, this porous structure is disordered, as evidenced by its small-angle XRD pattern (Fig. 1b). It can be clearly seen from Fig. 1b that there is only one broad peak at about 2° 2θ (Fig. 1b), which is a typical characteristic of a disordered framework.
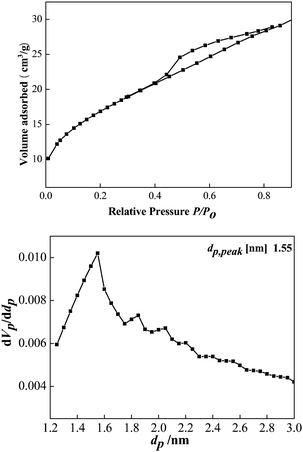 | ||
| Fig. 4 (a) Nitrogen sorption isotherm of the CeCu0.33 nanospheres; (b) the pore size distribution curves. | ||
It is well known that preparation methods often play a crucial role in improving the dispersion degree and the interfaces between particles, which greatly influence the catalytic performance.12 The exact role of short-periodic hydrothermal treatment used here was to achieve an unstable amorphous precursor with a larger portion of internal interfaces. The introduction of copper could hinder Ce species from segregating into the pure crystallites, which could maintain the desired high degree of interdispersion.6a,13 In addition, grain boundaries with a high degree of cleanness between CuO and CeO2 crystallites were formed due to incompatibility of lattice orientations between two neighboring crystal domains after calcination and consequently a portion of dispersed amorphous CuO caused phase separation.14 This fact may result in the formation of the unique porous structure, which makes a contribution to the enhanced catalytic activity for CO oxidation.
The porous CuO–CeO2 nanospheres were tested for CO oxidation under a reaction stream with a gas composition of 1.0 vol% CO balanced by fresh air with 20 vol% O2. Fig. 5a shows the CO conversion temperature on CuO–CeO2 catalysts with different amounts of CuO. It can be seen that the CeCu0.33 nanosphere catalyst exhibits the lowest complete conversion temperature (T100%) of CO oxidation as low as 71 °C. When the CuO amount was increased or decreased, such as to 0.46 and 0.22 or 0.28, they all exhibit a higher conversion temperature, 81, 84, and 93 °C. However, their catalytic performances are all far superior to those of CuO, CeO2, or a mechanical mixture of CeO2 and CuO. As clearly shown in Fig. 5b, CuO alone shows activity for CO oxidation, but the T100% is high (220 °C), while CeO2 alone and a mechanical mixture (molar ratio CeO2: CuO = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) show much higher T100% for CO oxidation under the same conditions at 490 and 698 °C, respectively. Obviously, the mechanical mixture is less active than as-prepared CuO–CeO2 catalysts or even pure CuO or CeO2. This fact indicates that there is a strong cooperative effect between CeO2 and CuO species in CuO–CeO2 nanocomposites. It is worth noting that CeCu0.33 catalyst has higher activity than the catalysts with higher or lower Cu content, although they exhibit same surface area.
:1) show much higher T100% for CO oxidation under the same conditions at 490 and 698 °C, respectively. Obviously, the mechanical mixture is less active than as-prepared CuO–CeO2 catalysts or even pure CuO or CeO2. This fact indicates that there is a strong cooperative effect between CeO2 and CuO species in CuO–CeO2 nanocomposites. It is worth noting that CeCu0.33 catalyst has higher activity than the catalysts with higher or lower Cu content, although they exhibit same surface area.
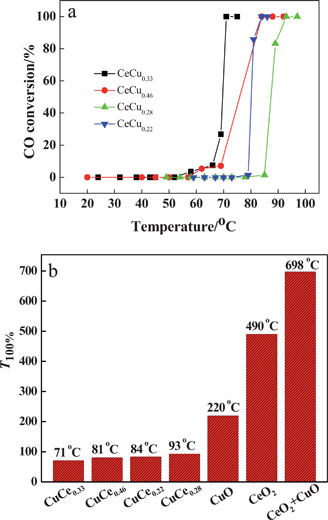 | ||
| Fig. 5 (a) CO oxidation temperature on various CuO–CeO2 catalysts; (b) CO oxidation temperature on CuO, CeO2, CuO–CeO2, and mechanical mixture of CeO2 and CuO. | ||
To further investigate the mechanism of CO oxidation on the as-synthesized CuO–CeO2 catalyst, surface analysis of the catalyst was carried out by X-ray photoelectron spectroscopy (XPS). We take the CeCu0.33 nanosphere sample as an example. The typical spectra of Ce 3d and Cu 2p binding energies of CeCu0.33 nanospheres are shown in Fig. 6. From Fig. 6a, it can be seen that the peaks centered at 899.2 and 917.2 eV could be ascribed to Ce4+ and those centered at 882.8 and 902.2 eV to Ce3+, suggesting that the Ce atom in the as-prepared CeCu0.33 nanospheres has two valence states.15 For Cu species, as shown in Fig. 6b, the presence of the shake-up peak and a higher Cu 2P3/2 binding energy (933.0–933.8 eV) indicates that the valence of Cu species is +2.12 Direct quantification of surface concentration ratio of Cu has been estimated by the integrated intensities of the Cu (2p) and Ce (3d) peaks confirmed by XPS (analysis depth of <2 nm).
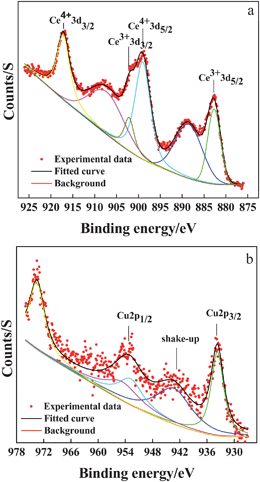 | ||
| Fig. 6 XPS spectra for as-prepared CeCu0.33 nanospheres: (a) Ce 3d; (b) Cu 2p. | ||
As-prepared CuO–CeO2 nanospheres have a surface Cu/(Cu + Ce) ratio of 13.62%. Compared with 33.44% for the nominal concentration confirmed by ICP, the surface concentration is lower than the nominal concentration, which indicates that there isn't enrichment of the surface with copper oxide species.12 The small size of CeO2 and low concentration CuO of the CuO–CeO2 surface may result in the high activity of the as-sythesized CuO–CeO2 nanospheres.
The enhancement in catalytic activity for the CeCu0.33 nanospheres could be further explained by H2 temperature-programmed reduction (H2-TPR). Here, we need to state in advance that pure CeO2 shows two reduction peaks, one at 528 °C, attributed to the reduction of surface oxygen species (capping oxygen), and the other at 820 °C, due to the reduction of bulk oxygen,16 while the as-synthesized nanospheres exhibit four major reduction peaks in the temperature range at 134.1 (α), 145.8 (β), 161.1 (γ), and 179.9 °C (δ), respectively, as shown in Fig. 7. This result is probably due to the as-synthesized sample with porous structure, higher surface area, and smaller crystallite size that was easier to reduce. The low temperature peak α is related to reduction of non-crystalline CuO strongly interacting with CeO2, while peak β is due to the reduction of larger CuO particles interacting weakly with CeO2. With respect to peak γ, it represents the reduction of crystalline forms of bulk CuO associated with CeO2 to some extent, while the reduction δ peak located at the highest temperature that represents the reduction of pure bulk CuO phase.17 In addition, the pure CuO bulk gets reduced at a high temperature of above 300 °C in the literature.18 From these reduction patterns, it can be concluded that the introduction of CuO species promotes the high activity of CeO2 because of the cooperative effect between them.
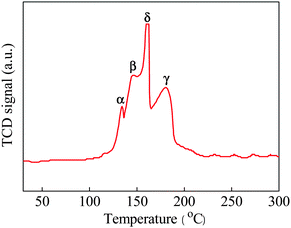 | ||
| Fig. 7 H2-TPR profile of as-prepared CeCu0.33 nanospheres. | ||
In this work, CO oxidation behavior is accepted as a suprafacial process which commonly takes place at interfaces between CuO and CeO2 nanoparticles. The surface lattice oxygen, as the oxidant, derives from the absorbed gaseous oxygen which is assumed to dissociate on the surface.19 The oxidant is usually surface lattice oxygen, and the oxidation of CO forming carbon dioxide leads to a mobile surface vacancy and a negative charge. The role of copper(II) and cerium(IV) is to eliminate the negative charges efficiently from synergism effect, and be reduced to copper(I) and cerium(III). In the literature, oxygen vacancies are created for charge balance around the Cu2+, which greatly occupying the surface Ce4+ sites lead to a large strain into the oxide lattice.4d,6a Vacancies can also strongly bind reactants and assist in their dissociation.20 Either lattice oxygen or dissociative dioxide absorbed at the surface reoxidates copper(I) and cerium(III) to the original valence, and replenishes the surface vacancy to regenerate the lattice oxygen sites.21 Ultimately, the CuO–CeO2 catalyst repeats absorption and oxidation of CO, then releases CO2 circularly.
4 Conclusions
In summary, the study reported here offers a simple synthesis route for synthesizing mesoporous CuO–CeO2 nanospheres with high surface area. In comparison to the corresponding materials obtained by conventional procedures, the as-prepared mesoporous CuO–CeO2 nanospheres exhibit enhanced low temperature catalytic property for CO oxidation. It is also believed that this established method can be expanded to obtain other mixed oxide nanostructures.Acknowledgements
This work was supported by the Natural Science Foundation of China (NSFC, Nos. 20731002, 10876002, 20771022 and 20973023), the 111 Project (B07012), Program for New Century Excellent Talents in University, Open Fund of State Key Laboratory of Explosion Science and Technology and Beijing Institute of Technology (No. ZDKT08-01 and YBKT09-13), Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP, No. 200800070015), and Funding Project for Science and Technology Program of Beijing Municipal Commission (No. Z09010300820902).References
- (a) M. Luo, J. Chen, J. Lu, Z. Feng and C. Li, Chem. Mater., 2001, 13, 1491 CrossRef; (b) A. I. Kozlov, D. H. Kim, A. Yezerets, P. Andersen, H. H. Kung and M. C. Kung, J. Catal., 2002, 209, 417 CrossRef CAS; (c) N. Sergent, J. Lamonier and A. Aboukaïs, Chem. Mater., 2000, 12, 3830 CrossRef CAS; (d) A. Bensalem, V. F. Bozon, M. Delamar and G. Bugli, Appl. Catal., 1995, 121, 81 CAS; (e) R. K. Usman, G. W. Graham, W. L. H. Watkins and R. W. McCabe, Catal. Lett., 1995, 30, 53 CrossRef; (f) J. Y. Luo, M. Meng, Y. Q. Zha and L. H. Guo, J. Phys. Chem. C, 2008, 112, 8694 CrossRef CAS.
- X. W. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746 CrossRef CAS.
- (a) W. H. Shen, X. P. Dong, Y. F. Zhu, H. R. Chen and J. L. Shi, Microporous Mesoporous Mater., 2005, 85, 157 CrossRef CAS; (b) M. F. Luo, J. M. Ma, J. Q. Lu, Y. P. Song and Y. J. Wang, J. Catal., 2007, 246, 52 CrossRef CAS.
- (a) G. Águila, F. Gracia and P. Araya, Appl. Catal., A, 2008, 343, 16 CrossRef CAS; (b) V. Ramaswamy, S. Malwadkar and S. Chilukuri, Appl. Catal., B, 2008, 84, 21 CrossRef CAS; (c) E. Moretti, M. Lenarda, L. Storaro, A. Talon, T. Montanari, G. Busca, E. Rodríguez-Castellón, A. Jiménez-López, M. Turco, G. Bagnasco and R. Frattini, Appl. Catal., A, 2008, 335, 46 CrossRef CAS; (d) X. Q. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martínez-Arias and M. Fernández-García, J. Phys. Chem. B, 2005, 109, 19595 CrossRef CAS; (e) A. Tschöpe, J. Markmann, P. Zimmer and R. Birringer, Chem. Mater., 2005, 17, 3935 CrossRef.
- B. Skårman, T. Nakayama, D. Grandjean, R. E. Benfield, E. Olsson, K. Niihara and L. R. Wallenberg, Chem. Mater., 2002, 14, 3686 CrossRef.
- (a) P. Bera, K. P. Priolkar, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro and N. P. Lalla, Chem. Mater., 2002, 14, 3591 CrossRef CAS; (b) W. J. Shan, W. J. Shen and C. Li, Chem. Mater., 2003, 15, 4761 CrossRef CAS.
- R. Kydd, W. Y. Teoh, K. Wong, Y. Wang, J. Scott, Q. H. Zeng, A. B. Yu, J. Zou and R. Amal, Adv. Funct. Mater., 2009, 19, 369 CrossRef CAS.
- P. Benito, M. Herrero, C. Barriga, F. M. Labajos and V. Rives, Inorg. Chem., 2008, 47, 5453 CrossRef CAS.
- (a) X. F. Chen, X. C. Wang and X. Z. Fu, Energy Environ. Sci., 2009, 2, 872 RSC; (b) J. Liu, M. Z. Li, J. X. Wang, Y. L. Song, L. Jiang, T. Murakami and A. Fujishima, Environ. Sci. Technol., 2009, 43, 9425 CrossRef CAS; (c) S. W. Liu, J. G. Yu and S. Mann, J. Phys. Chem. C, 2009, 113, 10712 CrossRef CAS; (d) W. Zhou, K. S. Liu, H. G. Fu, K. Pan, L. L. Zhang, L. Wang and C. Sun, Nanotechnology, 2008, 19, 035610 CrossRef.
- (a) S. N. Jacobsen, U. Helmersson, R. Erlandsson, L. R. Wallenberg and B. Skårman, Surf. Sci., 1999, 429, 22 CrossRef CAS; (b) L. Y. Kuo and P. Shen, Mater. Sci. Eng., A, 2000, 277, 258 CrossRef.
- Y. Liu, C. Wen, Y. Guo, G. Z. Lu and Y. Q. Wang, J. Phys. Chem. C, 2010, 114, 9889 CrossRef CAS.
- G. Avgouropoulos and T. Ioannides, Appl. Catal., B, 2006, 67, 1 CrossRef.
- M. Jobbágy, F. Mariño, B. Schönbrod, G. Baronetti and M. Laborde, Chem. Mater., 2006, 18, 1945 CrossRef CAS.
- A. Fuerte, R. X. Valenzuela and L. Daza, J. Power Sources, 2007, 169, 47 CrossRef CAS.
- (a) C. Q. Hu, Q. S. Zhu, Z. Jiang, Y. Y. Zhang and Y. Wang, Microporous Mesoporous Mater., 2008, 113, 427 CrossRef CAS; (b) Y. W. Zhang, R. Si, C. S. Liao and C. H. Yan, J. Phys. Chem. B, 2003, 107, 10159 CrossRef CAS.
- H. C. Yao and Y. F. Y. Yao, J. Catal., 1984, 86, 254 CrossRef CAS.
- X. C. Zheng, X. L. Zhang, X. Y. Wang, S. R. Wang and S. H. Wu, Appl. Catal. A, 2005, 295, 142 CrossRef CAS.
- X. Y. Jiang, G. L. Lu, R. X. Zhou, J. X. Mao, Y. Chen and X. M. Zheng, Appl. Surf. Sci., 2001, 173, 208 CrossRef.
- S. Royer, F. Bérubé and S. Kaliaguine, Appl. Catal., A, 2005, 282, 273 CrossRef.
- C. T. Campbell and C. H. F. Peden, Science, 2005, 309, 713 CrossRef CAS.
- P. G. Harrison, I. K. Ball, W. Azelee, W. Daniell and D. Goldfarb, Chem. Mater., 2000, 12, 3715 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |