Room temperature synthesis of highly hemocompatible hydroxyapatite, study of their physical properties and spectroscopic correlation of particle size†
Nagaprasad
Puvvada
,
Pravas Kumar
Panigrahi
and
Amita
Pathak
*
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur, 721302, West Bengal, India. E-mail: ami@chem.iitkgp.ernet.in
First published on 20th October 2010
Abstract
Needle shaped nanoparticles of hydroxyapatite (HA) have been synthesized at room temperature using orthophosphoric acid as the source of (PO4)3− ions, while calcium chloride, the calcium source, is suitably complexed with citric acid/tartaric acid/acetic acid. The presence of ligands inhibits the growth along [001] and [100] directions of the crystal and thus, helps in formation of needle shaped nanoparticles. The chemical compositions of the samples have been established through AAS and FTIR spectroscopy, while the crystallinity has been assessed through XRD and by the spectral changes in the υ1 and υ3 frequencies of the phosphate group in the respective FTIR spectra. The particle sizes of the samples have been determined from line broadening studies and correlations have been established between the curve fitted percentage area of FTIR and full width half height (FWHH) of the XRD peaks. TEM studies revealed the particle to be needle-shaped with a length and diameter in the range of 20–65 nm and 4–11 nm respectively. Changes in the surface charge of the water dispersed HA samples have been determined at different pH and the isoelectric point for the samples have been found in the range of 3.1–3.4. Finally, the morphology, surface area and hemocompatibility characteristics of the HA samples, prepared by using different complexing agents, have been compared.
Introduction
Hydroxyapatite (HA), a mineral with hexagonal symmetry, is the most stable form of calcium phosphate at room temperature and in the pH range of 4–12.1 There have been extensive efforts to synthetically produce HA in the nanocrystalline form since, it is the prototype of biological apatites that occur as the main inorganic component in bone, teeth and other calcified tissues.2 The properties of synthetically prepared HA have been reported to be influenced by the size and morphological characteristics of their particles.3 For example, smaller crystal size and more imperfect crystals, being subjected to dissolution,4,5 may affect the extent of bone loss in osteoporosis and other metabolic diseases.6 HA in the nanocrystalline form, enhances densification and improves the fracture toughness, and thus finds application as a bioactive and osteoconductive bone substitute material in clinical surgery. It is essential that the biomaterials used for drug delivery and biomedical application needed to be hemocompatible. This can be analyzed by hemolytic assay using rat red blood cells.7,8 Being a biocompatible material, they are promising drug delivery systems for the delivery of antitumor agents and antibodies in the treatment of cancer.9–11 HA has been used for the removal of numerous heavy metal ions (especially lead and cadmium ions) from waste water through ion-exchange process.12The method of preparation, along with the type of reagents used and control of the experimental parameters are known to affect the size and morphology of the particles, and consequently influence the properties of the final product. Therefore, HA sample with desired properties could be tailored through appropriate choice of the preparation route. Various methods that have been reported for the preparation of nanocrystalline HA include; chemical precipitation,13 in some cases followed by mechano-chemical,14,15 spray drying,16 electro-deposition,17 hydrothermal method,18 sol–gel,19 micro emulsion,20 wet chemical methods incorporating a freeze drying step21 and microwave irradiation.22 The wet chemical process based on precipitation route, is however the most convenient and commonly used process for the synthesis of nanocrystalline HA material. The process is simple, easy to use, and generates nanocrystalline HA at low temperatures. The process is also suitable for the precipitation of the appreciable quantities of apatites necessary for developing ceramic/ceramic, ceramic/metal and ceramic/polymer nanocomposites,23,24 which are widely used in medicine stomatalogy for repair of bone tissue.
In most of the conventional precipitation methods, the factors governing the precipitation, such as the pH, the temperature and the Ca/P mole ratio etc., are however not always precisely controlled, which results in the formation of HA nanocrystals with varying morphologies and sizes. Rodriguez-Lorenzo and Vallet-Regi25et al. have used the conventional precipitation method to produce nanosized HA powders with variable higher dimensions by using (NH4)2HPO4 as a phosphate source. Van Der Houwen et al. have synthesized calcium phosphate with citric acid and acetic acid as a complexing agent under titration method by using sodium hydrogen phosphate as a source of phosphate ion at pH 7.12 Based on the understanding that the grain-growth during the precipitation process could be controlled by capping the nuclei with organic complexing agents, Pramanik et al. synthesized nanoparticles of hydroxyapatite through chemical precipitation method by using various complexing agents like triethanolamine (TEA), ethylenediamine tetraacetic acid (EDTA), diethanolamine (DEA) and ethylene glycol (EG). They found that the smallest particles with 5–8 nm in diameter and 30–56 nm in length were formed, when TEA was used as capping agent, whereas the biggest particles with 12–16 nm in diameters and 80–120 nm in lengths were formed by using EG as capping agent.13 The specific surface areas for these particles ranged between 97 and 64 m2/gm respectively. The process is contributed by grain growth during the precipitation, which gives rise to particles with high aspect ratio. This problem can be overcome by arresting the growth of the nuclei using an organic complexing agent.26 Chemisorption of the organic ligands to the surface of the nucleating particles may inhibit the growth of the precipitating crystal in all the three dimensions but may, on the other hand, facilitate the preferential growth in one direction, consequently leading to formation of HA particles with higher aspect ratio (>4). Besides, the functional groups from the complexing agent may functionalize the surface of the nanocrystals, and make them more acceptable to the host tissues in biological applications. Hence, our aim is to prepare agglomerate free, less particle size of HA samples with high surface area.
The apatite crystal size has been determined by X-ray diffraction but directly cannot be applied to microscopic samples with rapid spatial variation in mineral structure such as biological tissue specimens. However, structural information of the biologically important apatite analogues, which are microscopic samples, can be readily obtained from IR spectroscopy. Here, we depict an IR-spectra based method to assess the crystallinity of HA mineral, based on the changes in the phosphate spectral region such as 900–1200 cm−1. This method can also be applied to the spectra of biologically applicable hydroxyl, carbonate, and fluoride substituted apatites.27 Fourier self-deconvolution, curve fitting and derivative spectroscopy technique was recently utilized to analyze the spectra of both synthetic as well as biological calcium phosphates.28,29 By using curve fitting, the percentage area of one of the subcomponents in the υ1 and υ3 phosphate (900–1200 cm−1) region is correlated with the changes in the particle size along the c-axis dimension which is measured through XRD line broadening analysis30 of the synthesized HA material.
In this paper, effort was made to synthesize nanosized HA particles through precipitation method starting from phosphoric acid and calcium chloride in presence of ammonia solution and different organic modifiers such as, acetic acid (AC), tartaric acid (TAT) and citric acid (CIT). Synthesized HA was characterized by FT-IR, XRD, SEM, HRTEM, BET, AAS, Zeta potential and Hemolytic assay. Further correlations between infrared subcomponents in the υ1 and υ3 phosphate region and changes in X- ray parameters were carried out. Finally, control growth mechanism was discussed for the prepared HA in presence of ligands.
Experimental section
Materials and methods
All the chemicals were used of analytical grade and available from commercial sources. CaCl2 (Merck Ltd, Mumbai, ≥ 98%), Glacial acetic acid (Merck Ltd, Mumbai, ≥99.5%) Tartaric acid (Merck Ltd, Mumbai, 99.7%), Citric acid (Merck Ltd, Mumbai, 98%) phosphoric acid (Merck (India) Ltd, Bombay, 85%) and ammonia (Merck Ltd, Mumbai, 25%) were used.Synthesis of hydroxyapatite nanoparticles
HA sample was synthesized by using calcium chloride and phosphoric acid as the source of calcium and phosphate ions respectively. Aqueous solution of calcium chloride (0.1M) and phosphoric acid (0.5 M) were mixed together as per required stoichiometry maintaining that Ca/P mole ratio is at 1.67. The pH of the resultant solution was maintained at 10.5 by adding concentrated ammonia to obtain white precipitate. It was then dried at 80 °C in a vacuum oven and grounded to fine powders and then characterized by different techniques. The resulted sample was named as HAP. The procedure for the preparation of HAP was further repeated using aqueous solution of chelated complexes in the starting solution by using three different ligands such as, acetic acid (AC), tartaric acid (TAT) and citric acid (CIT) and the resulted samples were respectively named as HAPAC, HAPTAT and HAPCIT. In the above synthesis procedures, the required stoichiometric ratios of calcium to ligand were maintained. A cloudy solution was obtained after about 5 min, when ammonia solution was added into the resultant solution, maintaining a pH around 10.5 (The organic modifiers were added to calcium ions solution before adding the phosphoric acid and ammonia solution in order to control the growth process).The possible overall chemical reaction involved in the process can be represented as follows:31
| 10Ca2+ + 6PO3−4 + 2 OH−→Ca10(PO4)6(OH)2 | (i) |
Eqn (i) may include the following set of intermediate reaction steps, shown by the eqn (ii)–(v).31–33
| CaCl2 + 2H2O→CaCl2·2H2O | (ii) |
| CaCl2·2H2O + X R–COOH→Ca(RCOO)X + 2Cl− + (X + 2)H+ + 2OH− | (iii) |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ca(R–COO)X + 6H3PO4 + 2OH−→6CaHPO4·2H2O + 4Ca2+(R–COO−)Y + (X − Y)R–COOH Ca(R–COO)X + 6H3PO4 + 2OH−→6CaHPO4·2H2O + 4Ca2+(R–COO−)Y + (X − Y)R–COOH | (iv) |
| 6CaHPO4·2H2O + 4Ca2+ + 8OH−→Ca10(PO4)6(OH)2 + 6H2O | (v) |
It is known that anhydrous calcium chloride reacts with water molecule to form hydrous calcium chloride (eqn (ii)). This hydrous calcium chloride then reacts with carboxylic acid groups, which is present in the organic modifiers used in the different reaction mixtures, to form calcium carboxylate complex according to eqn (iii). The reaction of Ca(R–COO)x with H3PO4 produces an water soluble compound, i.e., CaHPO4·2H2O, which is also reported as an intermediate in the modified Gee and Deitz method.32 The intermediate formed, in eqn (iv), finally generates HA in the presence of ammonium hydroxide (eqn (v)).
Characterization of nano sized hydroxyapatite
The functional group analysis in the as-dried powder of HA were carried out using Fourier transformed infrared (FTIR) spectroscopy (Perkin-Elmer Spectrum RXI instrument, within the scan range 4000–450 cm−1). Phase analysis of the powders were carried out by X-ray diffraction (XRD) using Cu-Kα radiation over 2θ range of 20°–60° at a scan rate of 1.1° min−1 and with a sampling interval of 0.02 at 30 mA and 40 kV by using Philips PW 1710 diffractometer. The powders aggregations in the samples were analyzed by digital imaging and scanning electroscope using the model JEOL JSM-5800 microscope at an accelerating voltage of 5 kV. The synthesized HA nanoparticles morphology and the particle size were measured by high-resolution transmission electron microscope (HRTEM) of JEOL JEM-2100 model with an acceleration voltage 200 kV. Particle size distribution was performed by a Laser 90 Plus particle size analyzer. Specific surface area measurements have been done by using Quantachrome Instruments (Autosorb-1, Model. No. ASI-C-9) BET surface area analyzer. Surface charge of the various samples has been investigated through zeta potential measurement using Zetasizer-4, Malvern instruments, U K. Atomic absorption Spectroscopy (AAS) has been performed to determine the concentration of calcium ion in HA samples by using Perkin Elmer, AAnalyst 700. Hemolytic activity was determined by measuring the absorption at 570 nm (Biorad Microplate reader 5804R).Results and discussion
In order to analyze the functional groups present in the various synthesized HA samples, FT-IR spectroscopy was carried out after overnight drying in vacuum oven at 80 °C. KBr pellets of the samples were prepared using 0.5 mg sample/100 mg KBr mixture by crushing and making translucent pellets in mechanical die press. Fig. 1. shows the FTIR spectra of different samples, recorded with in the scan range of 4000–450 cm−1. The observed bands were assigned to the corresponding possible functional groups and were listed in Table 1.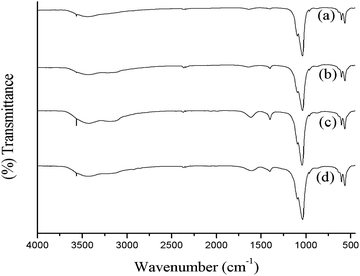 | ||
| Fig. 1 FTIR spectra of hydroxyapatite prepared (a) in the absence of ligand, with different ligands; (b) acetic acid (c) tartaric acid and (d) citric acid. | ||
| Assignment (hydroxyapatite) | Literature IR bands | HAP | HAPAC | HAPTAT | HAPCIT |
|---|---|---|---|---|---|
| O–H stretching | 3571 | 3566 | 3565 | 3565 | 3565 |
| 63433,34 | 635 | 634 | 635 | 637 | |
| PO–H stretching | 236535 | 2366 | 2364 | 2368 | 2367 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (carbonate ion) stretching O (carbonate ion) stretching |
139835 | 1400 | 1400 | 1406 | 1400 |
| PO stretching (υ3) | 109636 | 1094 | 1091 | 1091 | 1091 |
| PO stretching (υ3) | 103237 | 1037 | 1036 | 1034 | 1034 |
| PO stretching (υ1) | 96234–37 | 962 | 962 | 963 | 963 |
| PO stretching (υ2) | 47237 | 471 | 469 | 477 | 471 |
| PO bending (υ4′′′) | 56334,36 | 564 | 564 | 563 | 564 |
| PO bending (υ4′) | 60334,37 | 603 | 603 | 603 | 603 |
The phase analysis of hydroxyapatite nanoparticle was analyzed by XRD (Fig. 2.). All the XRD peaks were indexed to the lattice planes of hexagonal crystal structure with P63/m symmetry of synthesized HA nanoparticles with various ligands. The diffraction data are consistent with JCPDS file no. 09-432. The system has hexagonal symmetry and unit cell representation as depicted in schematic diagram in Fig S1.† The XRD peaks are broad in nature, which may be attributed to the lattice strain and low crystalline size of the formed apatite.38 The degree of crystallinity (Xc), corresponding to β002 is FWHH (°) of reflection (002) was evaluated by using the relation 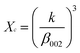 ; where k is constant and found to be 0.24 for a very large number of different HA powders.39 Crystallinity degree is decreased as shown in Table 2. The synthesized HA with various ligands nanoparticles have broader XRD peaks indicates nanocrystalline nature. The crystallite size has been calculated by using Scherrer's equation, i.e.
; where k is constant and found to be 0.24 for a very large number of different HA powders.39 Crystallinity degree is decreased as shown in Table 2. The synthesized HA with various ligands nanoparticles have broader XRD peaks indicates nanocrystalline nature. The crystallite size has been calculated by using Scherrer's equation, i.e. 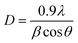 ; where D is the crystallite size, λ is the wavelength of the target material (1.5406 Å), β is full width at half height (FWHH) in radians, and θ is diffraction angle. The order of the crystalline size obtained using various ligands were found to be in the order: HAP > HAPAC >HAPTAT > HAPCIT. Hexagonal cell parameters (a and c) has been calculated by using the following relationship
; where D is the crystallite size, λ is the wavelength of the target material (1.5406 Å), β is full width at half height (FWHH) in radians, and θ is diffraction angle. The order of the crystalline size obtained using various ligands were found to be in the order: HAP > HAPAC >HAPTAT > HAPCIT. Hexagonal cell parameters (a and c) has been calculated by using the following relationship 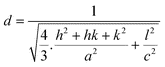 . The cell parameters are shown in Table 2.
. The cell parameters are shown in Table 2.
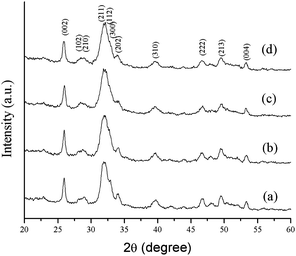 | ||
| Fig. 2 XRD patterns of the synthesized hydroxyapatite samples; (a) HAP (b) HAPAC (c) HAPTAT (d) HAPCIT. | ||
| Sample No. | Ligands used | Particle size/nm | Crystallinity (Xc) | Lattice parameters | |
|---|---|---|---|---|---|
| a | c | ||||
| 1 | None | 17–24 | 0.21 | 9.4156 | 6.8704 |
| 2 | AC | 15–21 | 0.18 | 9.4172 | 6.8527 |
| 3 | TAT | 13–17 | 0.15 | 9.4225 | 6.8436 |
| 4 | CIT | 11–15 | 0.13 | 9.4175 | 6.8592 |
Earlier X-ray diffraction technique was widely used as the main method for determination of apatite crystal size, while this method though directly cannot be applied for microscopic biological samples with spatial variation of crystal structure.30 The broad nature of this contour in biologic HA results from factors such as, vacancies in the HA lattice and lowering symmetry due to substitute ions either in the HA lattice or on the surface of the particles.28,29 Spectral changes of the phosphate contour in poorly crystalline biologic apatite's are characterized by determining the variations in the broad FT-IR contours of the poorly crystalline HA materials (Fig. 3.). Data reduction techniques such as Fourier deconvolution, curve fitting, and derivative spectroscopy were used for the determination of spectral structure correlation. Second derivative spectroscopy27 was utilized in the assignment of the factor group elements to peak positions in the υ1, υ3, and υ4 regions of HA. In view of the above drawback of XRD, IR method was developed40 to determine the percentage of cristallinity in apatite minerals at microscopic as well as macroscopic levels based on the changes in phosphate υ4 mode. In this method, the broad υ4 absorbance bands in 500–700 cm−1 region arise primarily from antisymmetric P–O bonding modes of the phosphate group. This mode, which is triply degenerate (F2 and Td symmetry) is resolved into at least two well defined peaks in HA materials.30 However, analysis of this spectral region does not give any determinative spectral-structural correlations.
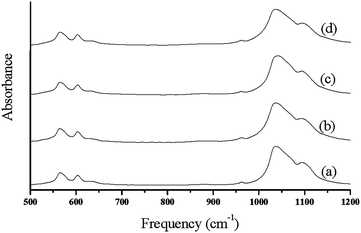 | ||
| Fig. 3 FT-IR spectra of synthesized hydroxyapatite nanoparticles; (a) HAP (b) HAPAC (c) HAPTAT (d) HAPCIT. | ||
IR method was used for the study of crystallinity of HA minerals, which is based on the changes in the phosphate υ1 and υ3 absorbance in 900–1200 cm−1. These spectral features arise primarily from the symmetric (υ1) and antisymmetric (υ3) P–O stretching modes of the synthesized HA phosphate groups.28 Therefore, we expect that the analysis of this spectral region would lead to multiple bands arising from both the phosphate υ1 and υ3 modes.27 Changes in this spectral region for a series of synthetic hydroxyapatites with crystalline sizes 6–20 nm were analyzed with curve fitting and second derivative spectroscopy. Analysis was done with peak fitting software and the peak-fitting algorithm creates Lorentzian-Gaussian bands that are added to produce a computed spectrum, which was compared with the experimental spectrum. To enhance the visibility of the changes in the FT-IR absorbance spectra accompanying maturation of the crystals, second-derivative spectroscopy was used to define the underlying bands in a better way. A mixed Gaussian-Lorentzian band shape was used to fit the region. Fig. 4. displays spectra representative of poorly crystalline HA, with the experimental and calculated contours overlaid along the individual sub-bands. It was reported that six or more components are necessary and sufficient for a suitable fit in this spectral region.27 To correlate the subtle changes in the FT-IR absorbance spectra with those of the XRD patterns, a curve fitting algorithm was used, the underlying components of both the FT-IR and X-ray data of synthesized HA was calculated. Fig. 5. depicts typical curve fit data of the FT-IR spectra and XRD patterns of all the poorly crystalline HA samples. For each of the HA synthesized samples, the crystal sizes calculated from the FWHH of the XRD peaks, after curve fitting were compared with the percentage areas of the FT-IR sub-bands. The percentage areas of the FT-IR sub-bands were plotted against the calculated crystal sizes using the FWHH of the corresponding X-ray diffraction peaks to obtain spectra-structure correlations as shown in Fig. 6.
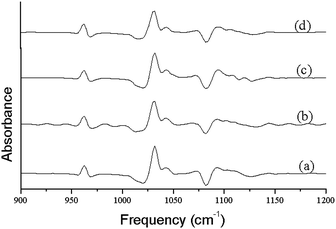 | ||
| Fig. 4 The second order derivative plots obtained from FT-IR spectra shown in Fig. 3. | ||
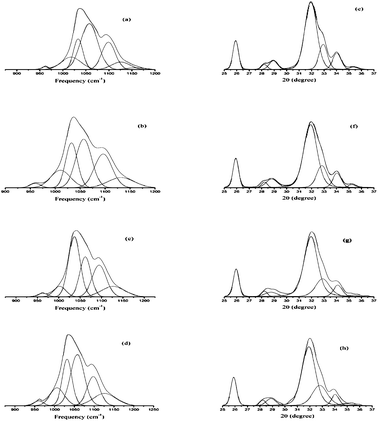 | ||
| Fig. 5 Curve fit of the FTIR spectra of the synthesized hydroxyapatite samples; (a) HAP, (b) HAPAC, (c) HAPTAT, and (d) HAPCIT and (e), (f), (g) and (h) represents the peak fit of X-ray diffraction patterns of the corresponding hydroxyapatite samples. | ||
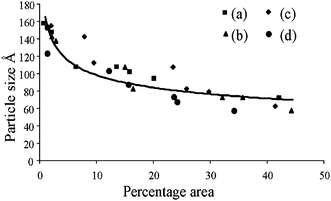 | ||
| Fig. 6 Relationship plot between crystalline size (from XRD patterns) and percentage area (from FT-IR spectra) of different hydroxyapatite samples; (a) without ligand, (b) acetic acid, (c) tartaric acid, (d) citric acid. | ||
FT-IR spectra of the synthesized nano hydroxyapatites, whose principal mineral components in each instance are poorly crystalline, agree extremely well with the applicability of the index of the crystallinity in biological materials.41 Nonlinear regression analysis led to the following relationship between fractional area of the FT-IR and crystalline size (Fig. 6.), calculated from the XRD, in the following relationship:
| Y = aXb + c (r = 0.87) |
| Y = Crystalline size (Å), X = Percent area, a = −28.03334, b = 0.3497, and c = 178.4 |
SEM micrographs (Fig S2†) reveal that the synthesized hydroxyapatite powders tend to aggregate in all the systems owing to the high surface energy of the nanosized particles. So the morphologies of the particles are unclear in the SEM micrographs. In the presence of the ligand, the aggregation tendency was found to decrease with increase in ligand coordination.
TEM studies were carried out for nanocrystalline HA samples and representative micrographs of HAP, HAPAC, HAPTAT and HAPCIT are shown in Fig. 7. The micrographs revealed the needle shaped morphology of the hydroxyapatite particles with highest degree of aggregation in these samples (which were prepared without using any ligand) while the aggregation was minimum for HAPCIT sample. The length and diameter of the particles of the different HA samples were determined from their respective micrographs, where the smallest visible aggregate was considered as a single particle. A summary of the particle dimension obtained from the TEM studies of the different hydroxyapatite samples were tabulated in Table 3. The corresponding selected area electron diffraction (SAED) patterns, shown in the insets of Fig. 7., indicate clearly visible rings, whose interplanar spacing are in good agreement with the characteristic spacing of apatite-like structure. On the basis of the aforementioned results, it is feasible to use the employed method for the preparation of needle-like HA particles with a high aspect ratio. The control of shape and morphology of these prepared samples is an actual challenge and further research is needed to validate the exact mechanism that determines nanocrystalline morphology.42 Pramanik et al. hypothesized that the morphology and shape of HA samples related to various complexing agents in which they were synthesized.13 From the morphological study of the particles, it was observed that HAPCIT sample depicted the highest aspect ratio among all the prepared samples while the HAPAC sample had the minimum value. The distribution in aspect ratio and average aspect ratio in each of the prepared HA samples are depicted in Fig. 8 (in this figure each particle is equal to average of ten particles aspect ratio). It was also observed that aspect ratio of the HAP sample was higher than the HAPAC sample, but in case of other ligands the aspect ratio was increased. It may be due to strong surface bound ligands but the particle size was decreased among all ligands. It is worthy to note that the diameters and lengths of the synthesized HA samples range from 3–11 nm and 20–65 nm, respectively. The difference in aspect ratios of these HA samples can satisfy the demands for potential applications in artificial bone and teeth.
 | ||
| Fig. 7 TEM micrographs of the synthesized hydroxyapatite samples; (a) HAP (b) HAPAC (c) HAPTAT (d) HAPCIT and the insets represent the corresponding SAED patterns. | ||
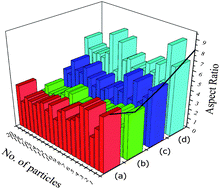 | ||
| Fig. 8 Histograms of average aspect ratios of different hydroxyapatite samples; (a), (b), (c) and (d) represents HAP, HAPAC, HAPTAT and HAPCIT respectively. | ||
The specific surface area of prepared HA samples were determined by nitrogen absorption analysis (BET) using a quanta chrome surface area analyzer. Samples were degassed at 150 °C for 4.5 h under vacuum prior to analysis. The BET surface area of the HAP, HAPAC, HAPTAT and HAPCIT powders were found to be 107.21, 121.26, 134.67 and 141.20 m2 g−1 respectively (Fig. 9). The corresponding average grain sizes of these HA were 17, 15, 14 and 13 nm as estimated by using the empirical equation d = 6/(ρS) (where d is the average grain size in micron, ρ is the density in g/cc and S is the specific surface area in m2 g−1).43 These values of grain sizes are in reasonable agreement with the results of the TEM observation and the average crystalline size estimated from the respective XRD peaks. The ligating ability of various complexing agents used in aqueous solution to form complex with the metal chloride may be responsible for the observed difference in the surface area of the synthesized samples. From the observations, it can be noted that as the density of carboxyl groups increases the yield of particle size decreases and surface area increases. However, we have also carried out a series of reactions to optimize the concentration of different ligands. However, the result of the optimization study has not been included in the present manuscript. After analyzing the morphological and surface area measurement results, we found that particles with minimum size and maximum surface area can be obtained by using the ligands at the concentrations mentioned in the paper.
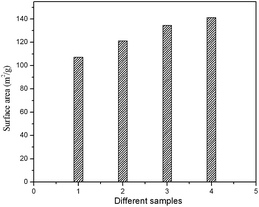 | ||
| Fig. 9 BET surface area of the prepared HA samples (1) HAP (2) HAPAC (3) HAPTAT (4) HAPCIT. | ||
The dried particles were dispersed in water to evaluate the zeta potential and particle size distribution (PSD). The particles showed negative potential at pH 10, the potential which increased with decreasing pH as shown in Fig. 10. The isoelectric point of the prepared HA samples are found to be in the range of 3.1–3.4. Zeta potential is very sensitive to the particle surface conditions, ions adsorbed on the particle surfaces, and the kind and concentration of ions in the solution. The variations in zeta potential are likely caused by difference in the surface conditions of the HA particles and interaction between particles and ions in the solution. The PSD analysis of the nanoparticles prepared with various ligands such as AC, TAT, and CIT shows substantial distribution of colloidal systems in PSD range of 110–225, 100–145 and 75–140 nm respectively (Fig S3†), while the HAP sample prepared without ligand shows PSD between 135–250 nm (Fig S3†). Further, the PSD analysis reveals the respective values of the mean particle sizes of HAP, HAPAC, HAPTAT and HAPCIT samples as 198, 155, 117 and 114 nm. All these values are found to be larger than those obtained from TEM observation. The aggregation of HA samples may be due to the relative bigger particle size observed under nanosizer. However, these aggregations were less pronounced in case of the nanoparticles prepared in the presence of ligands compared to that synthesized in the absence of any ligand. The details comparisons of particle sizes of synthesized samples from various measurements were incorporated in Table 3.
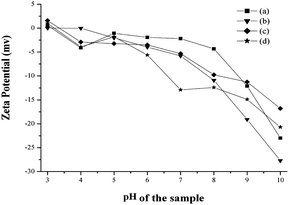 | ||
| Fig. 10 Zeta potential of hydroxyapatite samples; (a) HAP (b) HAPAC (c) HAPTAT (d) HAPCIT. | ||
Calcium ions content in the HA samples were determined using calcium ion calibration curve obtained from AAS. For the determination of calcium content by Flame Atomic Absorption Spectroscopy (FAAS) mode, the samples were dissolved in dilute HCl. The calibration curve was prepared over the concentration range of 0.1–30.0 μg ml−1 of calcium ions and the absorbance were measured at 422.7 nm beam produced by the hollow cathode lamp.
The mass fraction of calcium ions concentration in HAP samples were determined as 38.92% (HAP), 39.06% (HAPAC), 39.19% (HAPTAT) and 39.22% (HAPCIT). It should also note that the percent of mass fraction for all the prepared samples was increased in presence of complexing agents. It is due to strongly bound carboxylic groups to form complexes with the calcium ions.
The samples (HAP, HAPAC, HAPTAT, and HAPCIT) were dispersed in HEPES buffer to 10 mg ml−1 concentration. Hemocompatibility of the samples were analyzed using a protocol7 with some alteration. In brief, blood was obtained from 6-week-old BALB/c male mice and red blood cells (RBC) were collected by centrifugation (1500 g, 5 min, 4 °C) of the blood. The collected RBC pellet was diluted in 20 mM HEPES buffered saline (pH 7.4) to give a 5% (v/v) solution. The RBC suspension was added to HEPES-buffered saline containing 1% Triton X-100 and samples (HAP, HAPAC, HAPTAT, and HAPCIT) and incubated for 30 and 60 min at 37 °C. After completion of the incubation, all the control and samples were centrifuged (Heraeus table top centrifuge 5805R) at 12,000 rpm at 4 °C and the supernatants were transferred to a 96-well plate. Hemolytic activity was determined by measuring the absorption at 570 nm (Biorad Microplate reader 5804R). Control samples of 0% lysis (-ve control) (in HEPES buffer) and 100% lysis (+ve control) (in 1% Triton X-100) were employed in the experiment.44 All the assays were performed in triplicate. Hemolytic effect of each treatment was expressed as percent cell lysis relative to the untreated control cells (% control) defined as: [(Abs570 samples)/(Abs570 control cells)] × 100, where absorbance is abbreviated as Abs.
Previous report indicates that the percentage of hemolysis less than 5% and 5–10% are considered as highly hemocompatible and hemocompatible material respectively.45 It is found that all the synthesized nanoparticles showed significantly lower hemolytic activity (less than 5%) with respect to control samples (Fig. 11). But no significant difference was found in hemolytic activity among these samples. The high hemocompatibility of the synthesized hydroxyapatite nanoparticles can make them to be used for potential application in biomedical engineering. This high hemocompatibility of the nanoparticles may be attributed to the presence of surface hydrophilic groups (carboxy, hydroxyl functional groups).8
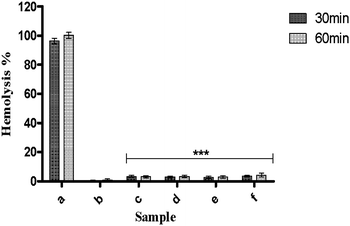 | ||
| Fig. 11 Hemolytic assay of samples; (c) HAP, (d) HAPAC, (e) HAPTAT, and (f) HAPCIT at 10 mg ml−1 concentration. (a) and (b) respectively represent +ve control 100% lysis (in 1% Triton X-100) and −ve control samples of 0% lysis (in HEPES buffer) employed in the experiment. The bars indicate the means ± SD (n = 3). Significant difference is shown as ***p < 0.001 versus +ve control. | ||
HRTEM analysis reveals particulars related to the growth mechanism that leads to formation of HA by controlling the growth in presence of various ligands. The dhkl values found from the FFT analysis agree with the reflection of HA. The most important surface, i.e., (001) is estimated through study of HRTEM. On the basis of measured reflections, it is envisaged that the control of the growth occurs along the (001) and (100) surface (Fig. 12) which is in agreement with recent study by Kwon et al.46 These face act as the binding site for many ionic species, including small molecules, polymers, and anionically modified cell surfaces. In this study, we have used literature which were reported on molecular modeling technique to study the binding of organic ligands on HA crystal surface, which eventually affects the crystal growth.47,48 The representative atomistic models of HA surfaces help in investigating optimized binding geometries and calculating binding energies by using parameters from the universal force field.48 The literature reported results confirm the preferential adsorption of ligands on the (001) and (100) surface.47–49 In order to have better understanding of the growth control of the particles, we have also prepared samples at a time interval of 6 h. The morphology (refer to the TEM images shown in Fig. S4†) and particle size (Table S1†) of the prepared samples is found to be comparable with those of the samples obtained after 5 min of reaction time as shown in Table 3. The data are supported by the zeta potential measurement, which shows nearly same zeta potential of the samples obtained after 5 min of reaction time (Fig. S5†). Thus, it can be said that the control of morphology may be due to the negatively charged carboxyl and hydroxyl groups present in the ligands, which are bound to calcium ions to form HA. Electrostatic interactions are therefore believed to occur between the cationic sites in the HA mineral and the anionic domains in the complexing agents. With the increasing densities of carboxyl groups, the abundant supply of coordination sites for complexation with calcium ions leads to a very large number of nuclei for the growth of HA particles, resulting in smaller crystal size.46 This may be the plausible elucidation for controlling the growth of the material. The proposed mechanism is also supported by the increase in the value of lattice parameter ‘a’ and decrease in the value of ‘c’ with reducing crystallite sizes.50 The high surface area and control of particle size have many more applications. For example, apoptotic study on cancer cells depends on the surface area and size particles.51
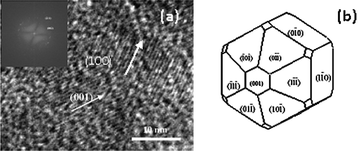 | ||
| Fig. 12 HRTEM image of HA prepared with citric acid ligand (a) (Inset: FFT image of corresponding HA), Schematic representations of estimated morphology of HA with ligands (SHAPE software) (b). | ||
Conclusion
In summary, we have successfully synthesized HA nanoparticles with high surface area by co-precipitation method using complexing agents. All the synthesized samples are characterized by various characterization techniques, such as XRD, TEM, PSD and SEM analysis. These analyses suggest that the synthesized hydroxyapatite powders under various complexing agents show smaller crystallites, particle sizes, less agglomeration compared to HA, synthesized in the absence of any ligand. Though, HRTEM images of the hydroxyapatite nanoparticles indicate the formation of acicular needle-like morphologies under all reaction conditions, the HA with complexing agents has shown a reduction in particle sizes. This observation has been attributed to the effective restriction of the nuclei growth (control the growth along the [001] and [100] axis) of the synthesized hydroxyapatite samples by ligands during precipitation process. No significant change in the particle size of HA is observed even after increasing reactive time interval. The correlation between infrared and X-ray parameters of these nanomaterials is found to be well matched. The synthesized HA nanoparticles shows highly hemocompatibility in bulk aqueous media, which depends on the shape of the particles (i.e., needle-like shape). The present study has provided not only novel building blocks for the construction of artificial bones with novel mechanical properties but also a new strategy for the controlled growth of inorganic nanoparticles. This work can further be extended for investigating the effect of high surface area of the HA nanoparticles on the antitumor activity, apoptosis and apoptotic signaling activation protein levels.Acknowledgements
Authors would like to acknowledge M.H.R.D., Govt. of India, for the financial support. The authors are grateful to Prof. M. Mahitosh for supporting the Hemolytic assay measurement, School of Medical Science and Technology, IIT-Kharagpur, India.Notes and references
- S. Koutsopoulos, J. Biomed. Mater. Res., 2002, 62, 600–612 CrossRef CAS.
- A. B. Sonju Clasen and I. E. Ruyter, Adv. Dent. Res., 1997, 11, 523–527 Search PubMed.
- R. Ramachandra, H. N Roopa and T. S. Kannan, J. Mater. Sci.: Mater. Med., 1997, 8, 511–518 CrossRef CAS.
- R. Z. LeGeros, Adv. Dent. Res., 1988, 2, 164–180 Search PubMed.
- S. Hurson, W. Lacefield, L. Lucas, J. Ong, R. Whitehead and J. Bumgardner, Transactions of the 19th Annual Meeting of the Society for Biomaterials, 1993Birmingham, AL, April 28–May 2, p. 223 Search PubMed.
- H. Glenn, Best Pract. Res. Clin. Rheumatol., 2008, 22, 1127–1139 CrossRef.
- K. Yasufumi, I. Tomoko, A. Tomohiro, S. Kosuke, K. Fumiaki, M. Noriyuki, Baba Kazuhiko and O. Naoto, Cancer Lett., 2008, 270, 260–268 CrossRef.
- Y. Zhilu, W. Jin, L. Rifang, F. M. Manfred, J. Fengjuan, S. Hong and H. Nan, Biomaterials, 2010, 31, 2072–2083 CrossRef CAS.
- W. Xia and J. Chang, J. Controlled Release, 2006, 110, 522–530 CrossRef CAS.
- Q. Yang, S. H. Wang, P. W. Fan, L. F. Wang, Y. Di, K. F. Lin and F. S. Xiao, Chem. Mater., 2005, 17, 5999–6003 CrossRef CAS.
- Y. Yamashita, A. Uchida, T. Yamakawa, Y. Shinto, N. Araki and K. Kato, Int. Orthop., 1998, 22, 247–251 CrossRef CAS.
- J. A. M. Van Der Houwen and E. Valsami-Jones, Environ. Technol., 2001, 22, 1325–1335 CrossRef CAS.
- P. Nabakumar, T. Abhijit and P. Panchanan, J. Mater. Process. Technol., 2007, 184, 131–138 CrossRef.
- K. C. B. Yeong, J. Wang and S. C. Ng, Biomaterials, 2001, 22, 2705–2712 CrossRef CAS.
- C. Mochales, H. E. Briak-Benabdeslam, M. P. Ginebra, A. Terol, J. A. Planell and P. Boudeville, Biomaterials, 2004, 25, 1151–1158 CrossRef CAS.
- P. Luo and T. G. Nieh, Biomaterials, 1996, 17, 1959–1964 CrossRef CAS.
- M. Shirkhanzadeh, J. Mater. Sci.: Mater. Med., 1998, 9, 67–72 CrossRef CAS.
- J. S. Earl, D. J. Wood and S. J. Mile, J. Phys. Conf. Ser., 2006, 26, 268–271 CrossRef.
- I. Bogdanoviciene, A. Beganskiene, K. Tonsuaadu, J. Glaser, H. J. Meyer and A. Kareiva, Mater. Res. Bull., 2006, 41, 1754–1762 CrossRef CAS.
- G. K. Lim, J. Wang, S. C. Ng, C. H. Chew and L. M. Ganl, Biomaterials, 1997, 18, 1433–1439 CrossRef CAS.
- M. J. Philips, J. A. Darr, Z. B. Luklinska and I. Rehman, J. Mater. Sci.: Mater. Med., 2003, 14, 875–882 CrossRef CAS.
- L. Jingbing, L. Kunwei, W. Hao, Z. Mankang and Y. Hui, Chem. Phys. Lett., 2004, 396, 429–432 CrossRef.
- R. Morrissey, L. M. Rodriguez-Lorenzo and K. A. Gross, J. Mater. Sci.: Mater. Med., 2005, 16, 387–392 CrossRef CAS.
- W. Jie, L. Yubao, C. Weiqun and Z. Yi, J. Mater. Sci., 2003, 38, 3303–3306 CrossRef.
- L. M. Rodriguez-Lorenzo and M. Vallet-Regi, Chem. Mater., 2000, 12, 2460–2465 CrossRef CAS.
- F. C. Meldrum, N. A. Kotov and J. H. Fendler, Langmuir, 1994, 10, 2035–2040 CrossRef CAS.
- S. J. Gadaleta, E. P. Paschalis, N. P. Camacho, F. Betts, R. Mendelhson and A. L. Boskey, Mineral scale formation and inhibition,New York, Plenum Press, 1995, 283–294 Search PubMed.
- C. Rey, M. Shimuzu, B. Collins and M. J. Glimcher, Calcif. Tissue Int., 1991, 49, 383–388 CAS.
- C. Rey, M. Shimuzu, B. Collins and M. J. Glimcher, Calcif. Tissue Int., 1990, 46, 384–394 CAS.
- N. Pleshko, A. Boskey and R. Mendelsohn, Biophys. J., 1991, 60, 786–793 CrossRef CAS.
- W. K. Dong, C. In-Sun, Y. K. Jin, L. J. Hae, S. H. Gill, R. Hyun-Seung, S. Heungsoo, S. J. Hyun, K. Hyungtak and S. H. Kug, Langmuir, 2009, 26, 384–388.
- A. Gee and V. R. Deitz, J. Am. Chem. Soc., 1955, 77, 2961–2965 CrossRef CAS.
- M. Markovic, B. O. Fowler and M. S. Tung, J. Res. NIST, in preparation.
- Z. Yanjie and L. Jinjun, Cryst. Growth Des., 2008, 8, 2101–2107 CrossRef CAS.
- H. Eshtiagh-Hosseini, M. R. Houssaindokht, M. Chahkandhi and A. Youssefi, J. Non-Cryst. Solids, 2008, 354, 3854–3857 CrossRef CAS.
- A. Slosarczyk, Z. Pasziewicz and C. J. Paluszkiewicz, J. Mol. Struct., 2005, 744–747, 657–661 CrossRef.
- A. Rapacz-Kmita, C. Paluszkiewicz, A. Slosarczyk and Z. Pasziewicz, J. Mol. Struct., 2005, 744–747, 653–656 CrossRef CAS.
- R. Murugan and S. Ramakrishna, Cryst. Growth Des., 2005, 5, 111–112 CrossRef CAS.
- E. Landi, A. Tampieri, G. Celotti and S. Sprio, J. Eur. Ceram. Soc., 2000, 20, 2377–2387 CrossRef CAS.
- J. D. Termine and A. S. Posner, Nature, 1966, 211, 268–270 CrossRef CAS.
- R. Z. LeGeros, Prog. Cryst. Growth Charact., 1981, 4, 1–45 CAS.
- B. Susmita and K. S. Susanta, Chem. Mater., 2003, 15, 4464–4469 CrossRef CAS.
- L. Zhongjun and W. Peiyuan, J. Mater. Sci., 2005, 40, 6589–6591 CrossRef.
- D. Guggi, N. Langoth, M. H. Hoffer, M. Wirth and A. Bernkop-Schnürch, Int. J. Pharm., 2004, 278, 353–360 CrossRef CAS.
- P. Nabakumar, M. Sasmita, A. Sarfaraz and P. Panchanan, Polym. Compos., 2008, 29, 429–436 Search PubMed.
- K. Ki-Young, W. Eddie, C. Alice, C. Neil, S. Eduardo, C. Uh-Joo, K. Maxwell and L. Seung-Wuk, Langmuir, 2008, 24, 11063–11066 CrossRef.
- N. Almora-Barrios, K. F. Austen and N. H. de Leeuw, Langmuir, 2009, 25, 5018–5025 CrossRef CAS.
- R. T. Zhengrong, A. V. James, L. Jun, M. Bonnie, J. M. Matthew, A. R. Mark, K. Hiromi and X. Huifang, Nat. Mater., 2003, 2, 821–826 CrossRef CAS.
- Haihua Pan, Jinhui Tao, Xurong Xu and Ruikang Tang, Langmuir, 2007, 23, 8972–8981 CrossRef CAS.
- B. Sujin, G. Jean-Christophe and N. Taco, J. Chem. Phys., 2009, 130, 064504 CrossRef.
- Y. Yuan, L. Changsheng, Q. Jiangchao, W. Jing ang and Z. Yuan hang, Biomaterials, 2010, 31, 730–11, DOI:10.1016/j.biomaterials.2009.09.088.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S5. See DOI: 10.1039/c0nr00611d |
| This journal is © The Royal Society of Chemistry 2010 |