Synthesis of core-shell gold coated magnetic nanoparticles and their interaction with thiolated DNA†
Ian
Robinson
a,
Le D.
Tung
b,
Shinya
Maenosono
c,
Christoph
Wälti
d and
Nguyen T. K.
Thanh
*ef
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
bDepartment of Physics, University of Liverpool, Crown Street, Liverpool, L69 7ZE, UK
cSchool of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan
dSchool of Electronic and Electrical Engineering, University of Leeds, Leeds, LS2 9JT
eThe Davy-Faraday Research Laboratory, The Royal Institution of Great Britain, 21 Albemarle Street, London, W1S 4BS, UK
fDepartment of Physics & Astronomy, University College London, Gower Street, London, WC1E 6BT, UK. E-mail: ntk.thanh@ucl.ac.uk; Fax: +44 2076702920; Tel: +44 2074916509
First published on 21st October 2010
Abstract
Core-shell magnetic nanoparticles have received significant attention recently and are actively investigated owing to their large potential for a variety of applications. Here, the synthesis and characterization of bimetallic nanoparticles containing a magnetic core and a gold shell are discussed. The gold shell facilitates, for example, the conjugation of thiolated biological molecules to the surface of the nanoparticles. The composite nanoparticles were produced by the reduction of a gold salt on the surface of pre-formed cobalt or magnetite nanoparticles. The synthesized nanoparticles were characterized using ultraviolet-visible absorption spectroscopy, transmission electron microscopy, energy dispersion X-ray spectroscopy, X-ray diffraction and super-conducting quantum interference device magnetometry. The spectrographic data revealed the simultaneous presence of cobalt and gold in 5.6 ± 0.8 nm alloy nanoparticles, and demonstrated the presence of distinct magnetite and gold phases in 9.2 ± 1.3 nm core-shell magnetic nanoparticles. The cobalt-gold nanoparticles were of similar size to the cobalt seed, while the magnetite-gold nanoparticles were significantly larger than the magnetic seeds, indicating that different processes are responsible for the addition of the gold shell. The effect on the magnetic properties by adding a layer of gold to the cobalt and magnetite nanoparticles was studied. The functionalization of the magnetic nanoparticles is demonstrated through the conjugation of thiolated DNA to the gold shell.
Introduction
Magnetic nanoparticles (NPs) are of importance to various biomedical applications such as magnetic separation, cell labelling, targeted drug delivery, hyperthermia treatment of solid tumours and contrast agents for magnetic resonance imaging (MRI).1–5 In most cases, it is important to control accurately the size, shape and composition of the NPs. This can commonly be achieved through methods based on the thermal decomposition of organometallic complexes in the presence of hydrophobic ligands such as oleic acid, trioctyl phosphine oxide and oleyl amine.6–9 This, however, produces NPs that cannot be easily dispersed in water. One method to overcome this challenge is the post-synthesis modification of the surface of the NPs to produce a core-shell system.The versatility of core-shell NPs compared to those fabricated from a single source also makes them an increasingly interesting subject of research. Core-shell magnetic NPs comprise a magnetic core (e.g. cobalt, iron oxide, etc) and a shell that can provide not only a hydrophilic layer to the NPs but also a platform for the surface functionalization of the NPs. The shell can be generated by replacing the hydrophobic ligand with macromolecules such as peptides10,11 and hydrophilic polymers12–16 or other ligands such as DMSA (2,3-dimercaptosuccinic acid),17 betaine hydrochloride18 and silanes.19 Alternatively, a gold salt can be reduced at the NP surface to add a Au layer,20–25 which can then be functionalized with thiolated molecules via a covalent bond to the surface of the NP.
There are several examples reporting the synthesis of Au coated magnetic NPs. For example, Lee et al.21 coated 6.5 nm Co NPs with a Au shell. This was achieved by heating the Co particles in 1,2-dichlorobenzene, under reflux, with [(C8H17)4N]+[AuCl4]− containing trioctylphosphine (TOP) as a stabilizer. A by-product of the reaction was analyzed and found to be CoCl2, indicating the core-shell structure was formed by a process of redox transmetallation between Co0 and Au3+. Furthermore, the core-shell structure of the NPs was verified by high resolution TEM (HRTEM). Co NPs have also been coated with Au by a chemical reduction method. The Co particles were synthesized using 3-(N,N-dimethyldodecylammonio)propanesulfonate as the stabilizer and lithium triethylhydridoborate as the reducing agent. Using ultrasonication in an inert atmosphere, a solution of KAuCl4 in tetrahydrofuran was used to form the gold shell via reduction of the Au3+ ions on the Co nanoparticle surface.26 A similar process led to the formation of Au-coated iron NPs that are stable under neutral and acidic aqueous conditions.27 FeCl3, dissolved in N-methyl-2-pyrrolidone (NMP), was reduced by sodium to form the metallic core with benzylpyridine as the capping ligand. The Au layer was deposited by the addition of HAuCl4, which was dissolved in NMP. This type of core-shell nanoparticle can also be prepared by a reverse microemulsion method. The inverse micelles were formed with cetyltrimethylammonium bromide as the surfactant, 1-butanol as a co-surfactant and octane as the continuous oil phase. FeSO4 was then reduced using NaBH4, followed by the addition of HAuCl4 to coat the iron particles.28
Here, we report the synthesis and characterization of Co-Au and Fe3O4-Au NPs that were prepared by reducing a gold (III) salt in the presence of the respective magnetic NPs. The Co and Fe3O4 NPs were prepared in organic solvent using standard synthesis methods and coated with sodium bis(2-ethylhexyl) sulfosuccinate (NaAOT) and oleic acid, respectively. A Au layer was subsequently added using previously published methods.20,29 However, we do present a novel technique for transferring the Au coated magnetic NPs into aqueous solution and their subsequent bio-functionalization. The presence of the Au shell on the magnetic core makes it possible to functionalize the NPs with thiolated molecules by exploiting the Au-S chemistry. In particular, it facilitates the attachment of biological molecules with inherent self-assembly properties onto the surface of NPs. This opens up new ways for assembling magnetic NPs rationally into well-organized and functional complexes through the lock-and-key functionality provided by the biological molecules on the surface. Here, we functionalize the magnetic core-shell particles with 11-mercaptoundecanoic acid and thiolated single-stranded DNA as a proof-of-principle. Ultraviolet-visible (UV-vis) absorption spectroscopy and X-ray diffraction (XRD) were used to detect the presence of Au in the NPs and transmission electron microscopy (TEM) and HRTEM were employed to study the morphology of the Au coated NPs. The effects on the magnetic properties of the NPs by the addition of Au were investigated using a super-conducting quantum interference device (SQUID) magnetometer.
Experimental section
Chemicals
Cobalt octacarbonyl (Co2(CO)8, 99.9%), iron(III) acetylacetonate (Fe(acac)3, 99.9%), sodium bis(2-ethylhexyl) sulfosuccinate (NaAOT, 99%), hydrogen tetrachloroaurate (III) hydrate (HAuCl4·3H2O, 99.9%), 1,2-hexadecanediol, (90%), oleylamine (70%), oleic acid (99%), phenyl ether (99%), mercaptoundecanoic acid (95%), and other solvents (toluene and ethanol) were purchased from Sigma-Aldrich (Dorset, UK). Gold (III) acetate (Au(ac)3, 99.9%) was purchased from Alfa Aesar. All chemicals were used as received.Synthesis of NaOAT coated Co NPs
A solution of Co2(CO)8 (0.49 g, 1.43 mmol) in anhydrous toluene (3 ml) was rapidly injected into a solution of NaAOT (0.067 g, 0.151 mmol) in toluene (27 ml) at 110 °C under N2. The solution was vigorously stirred for 6 h at this temperature and then cooled to room temperature.Synthesis of Co–Au NPs
A 5 ml aliquot of the toluene solution of NaAOT coated Co NPs was diluted with toluene (20 ml) and heated to 85 °C. A solution of HAuCl4·3H2O (0.05 g) and oleylamine (1.25 ml) in toluene (5 ml) was injected into the Co solution and the temperature was maintained at 85 °C for 1 h then cooled to room temperature to produce a dark purple solution.A permanent magnet was placed on the outside of a glass vial containing the Co–Au NPs to attract the magnetic material to the side of the vial. The supernatant was removed and the remaining magnetic fraction was resuspended in toluene. This magnetic separation procedure was repeated a further two times and all characterizations were carried out on this fraction.
Synthesis of Fe3O4 NPs
Iron acetylacetonate (0.71 g, 2 mmol) was dissolved in phenyl ether (20 mL) with oleic acid (2 mL, 6 mmol) and oleylamine (2 mL, 4 mmol) under N2 with vigorous stirring. 1,2-Hexadecanediol (2.58 g, 10 mmol) was added into the solution and it was heated under reflux for 2 h, then cooled to room temperature. The solution was used as prepared without any further separation.Synthesis of Fe3O4-Au NPs
The Fe3O4 NPs (10 mL of phenyl ethyl solution, approximately 0.33 mmol Fe3O4), Au(ac)3 (0.83 g, 2.2 mmol), 1,2-hexadecanediol (3.1 g, 12 mmol), oleic acid (0.5 mL, 1.5 mmol) and oleylamine (3 mL, 6 mmol) were added to phenyl ether (30 mL) under N2 flow with vigorous stirring. The reaction solution was heated to 180–190 °C and held at this temperature for 1.5 h.After cooling to room temperature, ethanol was added into the solution and the magnetic material collected using a permanent magnet. The precipitated product was washed with ethanol, and redispersed in hexane (10 mL) in the presence of oleic acid and oleylamine (approx 75 mM of each). The NP solution appeared dark purple.
Phase transfer of Fe3O4-Au NPs
The Fe3O4-Au NPs (5 ml of hexane solution) were precipitated in ethanol (approx 15 ml) and by a permanent magnet. The precipitate was washed two more times then redispered in 3 ml of 1 M TMAOH solution. Tri-sodium citrate (0.04 g) was added and the pH of the resulting solution adjusted to pH ≈ 6.5. The solution was sonicated for 15 min, after which the Fe3O4-Au NP were collected using a magnet and redispersed in pure water (5 mL) and sonicated for a further 5 min in a Fisher Scientific ultrasonic bath FBE#FB1505 using sweep mode at room temperature. An aliquot of this solution (100 μL) was then added to 100 μl of thiolated DNA solution (100 μM, single stranded, 20 nucleotide long, 5′ thiolated via C6 linker, purchased from Eurofins) and the solution was observed for any indication of aggregation. One nucleotide unit is 0.34 nm long, therefore the length of DNA added is about 7 nm.Characterization
The UV/visible spectra were obtained using a Molecular Devices Spectromax 384 spectrometer. TEM images were obtained using an FEI Tecnai G2 120 kV TEM operated at 100 kV and visualized using analySIS software. HRTEM images were obtained using a Hitachi H-9000NAR H with 300 kV acceleration voltage, operated at 300 kV. The diameter (d) of the NPs was taken as the mean of a minimum of 200 (n) NPs measured using Bersoft Image Measurement 5.2 software. The magnetic properties of the NPs, including hysteresis, zero field cooled (ZFC) and field cooled (FC) response curves, were obtained using a Quantum Design Magnetic Property Measurement System (MPMS) XL SQUID magnetometer. XRD patterns were obtained using a Rigaku RINT-2500 diffractometer (CuK radiation line λ = 1.5408 Å; 40 kV/100 mA).Results and discussion
Cobalt and iron oxide NPs were synthesized in organic solvent using standard methods and coated with NaAOT and oleic acid, respectively. This was followed by the reduction of a gold salt in order to add a layer of gold on the Co and iron oxide NPs. It can be seen from the TEM images in Fig. 1 that both the NaAOT coated Co and Co–Au NPs are spherical with a narrow size distribution (standard deviation of 0.8 nm for both types of NPs). Both the Co and the Co–Au NPs have similar average diameters as would be expected for an atom exchange process, where for each Au3+ ion reduced, a Co0 atom is oxidized to Co2+, resulting in no overall change in NP diameter.21 This suggests that the NPs were formed through a redox transmetallation process rather than a deposition process such as galvanization.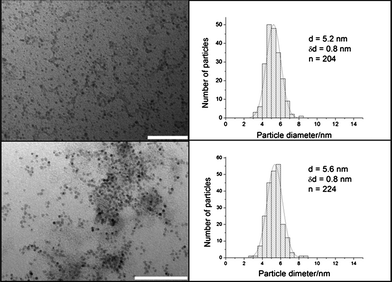 | ||
| Fig. 1 TEM images and size distributions of (a) NaAOT coated Co NPs and (b) Co–Au NPs. There is no significant difference in the size between the two types of NPs. Scale bar 100 nm. | ||
A HRTEM image and an energy dispersion X-ray (EDX) spectrum of the Co–Au NPs are depicted in Fig. 2. The spectrum shows peaks at 2.5 and 9.5 MeV that correspond to Au and a peak at 7.0 MeV which corresponds to Co, suggesting that the NPs have a Co–Au bimetallic structure. HRTEM shows that there is no distinct separation between the Co and Au phases, suggesting the formation of a Co–Au alloy.30 Moreover, it has been suggested that NPs produced by a similar method have slightly more complicated structure than a simple core-shell. Cheng et al.31 suggested that Au not only grows on the surface of the NPs, but will also diffuse into the Co cores, to produce metastable Co–Au NPs.
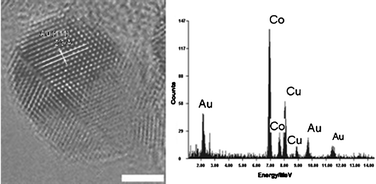 | ||
| Fig. 2 HRTEM image and EDX spectrum of a Co–Au magnetic NP. Scale bar 2 nm. | ||
The crystal lattice appears to be distorted on the nanoparticle surfaces because of a curvature effect. Therefore, the shell material, having a large lattice mismatch, can be epitaxially grown on the surface of core. This is completely different when the growth is on a flat surface. It is therefore difficult to define clearly the structure as core-shell or alloy, when the shell thickness is thin. Nonetheless, the lattice spacing estimated from the HRTEM image is 0.22 nm. This value is in between the Au (111) d-spacing (0.236 nm) and the Co (111) d-spacing (0.2045 nm). This result is consistent with the bimetallic alloy formation, i.e. the Co lattice fringe is slightly expanded due to the Au incorporation. It is important to note that alloying between Co and Au seems to take place only near the surface of Co at the interface between Co and Au.
Attempts to transfer these NPs to aqueous solution using 11-mercaptoundecanoic acid as a stabilizing ligand resulted in agglomeration and precipitation of the NPs. This may be a consequence of the rather short length of the incoming ligands, which do not effectively form a protective layer and do not provide the same degree of steric stabilization compared with the oleylamine. In addition, the low gold content on the surface of the NPs further hinders strong binding of the ligands.
In contrast to the Co–Au versus Co NPs, the average diameter of the Fe3O4-Au NPs is increased to 9.2 ± 1.3 nm from 6.7 ± 0.7 nm for the Fe3O4 seeds upon the reduction of gold acetate (Fig. 3a and b). This suggests that a Au layer with an average thickness of ∼1.2 nm was added to the surface of the NPs, resulting in an increase in diameter. The hydrophobic ligands (oleic acid and oleylamine) were removed from the surface of Fe3O4-Au NPs which were then stabilized with citrate and dispersed in water. The water dispersed Fe3O4-Au NPs could then be functionalized with thiolated DNA. The thiolated DNA functionalized Fe3O4-Au NPs have a negatively charged surface, owing to the exposed phosphate groups of the DNA, which enhances the stability of NPs in aqueous solution. Fig. 4 shows the HRTEM image and EDX spectrum of a single Fe3O4-Au NP. The spectrum has peaks at 0.75 MeV, 6.5 MeV and 7 MeV that correspond to metallic Fe and peaks at 2 MeV, 9.5 MeV and 11.5 MeV for Au. Furthermore the HRTEM image shows variation in contrast between the dark Fe3O4 core and the lighter Au shell. The core appears darker on this image as mass contrast appears to dominate over diffraction contrast, rendering the shell lighter even though Au has a higher electron density than Fe3O4. The lattice distances measured for the shell correspond to the known Au lattice parameters for the (111) phase and those measured for the core match well with the Fe3O4 lattice parameters for the (311) phase.
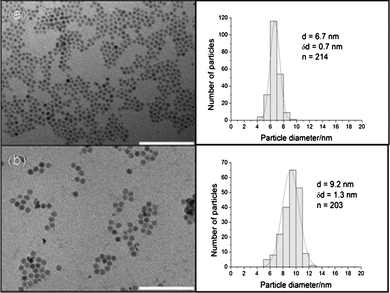 | ||
| Fig. 3 TEM images and size distributions of (a) Fe3O4 NPs in hexane and (b) Fe3O4-Au NPs in water. The Fe3O4-Au NPs are approximately 2.5 nm larger than the Fe3O4 NPs. Scale bar 100 nm. | ||
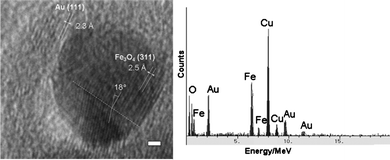 | ||
| Fig. 4 HRTEM image and EDX spectrum of a Fe3O4-Au NP. Scale bar 1 nm. | ||
Twinning in the Fe3O4 core crystal can be seen in Fig. 4, with the lattice spacing being the same on both sides of the twin boundary. The angle between the two separate crystals is consistent with previously observed values in twinned NP. Twinning occurs when two crystals of the same type intergrow with only a slight misalignment between them and atoms are shared between the two crystals at regular intervals. This results in a much lower-energy interface compared to grain boundaries that form when crystals of arbitrary orientation grow together. Although the XRD data cannot unambiguously determine whether the crystal is Fe3O4 or γ-Fe2O3, closer inspection of the position of each peak suggests the crystal to be predominately Fe3O4.
Fig. 5 shows the XRD pattern of Co–Au NPs having diffraction peaks at 2θ = 38.2°, 44.4°, 65.6° and 77.5°, which can be indexed to (111), (200), (220) and (311) planes of Au in the cubic phase. The absence of any diffraction peaks for Co is probably due to the heavy atom effect of the Au32 as it combines with the Co NPs. This effect has been previously observed in similar NPs.20,33,34
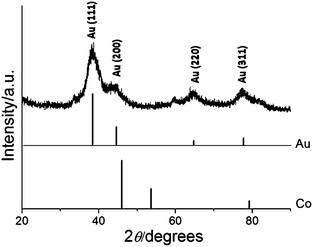 | ||
| Fig. 5 XRD pattern of Co–Au NPs with reference peaks for Co and Au. | ||
The XRD patterns of Fe3O4 and Fe3O4-Au NPs are compared in Fig. 6. A closer look at each peak reveals that the peak positions of the iron oxide are closer to those of Fe3O4 phase than those of γ-Fe2O3 phase. The XRD pattern of Fe3O4-Au NPs (curve b) displays peaks for both Fe3O4 and Au, however the peaks for the magnetite are subdued when compared to the XRD pattern of Fe3O4 alone (curve a). This is most likely caused by the heavy atom effect from Au32 as a result of the formation of Au-coating on the Fe3O4 NPs. As mentioned above this effect has been observed previously20,33,34 and provides further evidence for the presence of Au in the NPs.
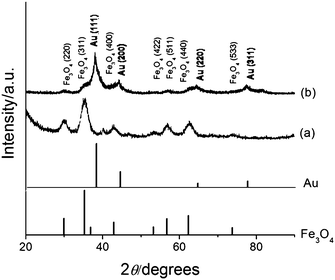 | ||
| Fig. 6 XRD patterns of (a) Fe3O4 and (b) Fe3O4-Au NPs with reference peaks for Fe3O4 and Au. | ||
The average crystalline size of the Fe3O4 core particles was calculated by the Scherrer formula using the half-maximum full width of the Fe3O4 (311) peak and found to be 5.2 nm, which is slightly smaller than, but in close agreement with, the 6.7 nm derived from the TEM images in Fig. 3. This presents a very interesting NP system as the core is very small, which would allow certain applications where the overall size of NPs is restricted, for example owing to the requirement of lower degrees of opsonization and for efficient trans membrane permeation and excretion in the biological system.35 The small NPs are also well suited for the decoration of nanoscale biological scaffolds such as DNA.
The UV/visible absorption spectra for Co, Au and Co–Au bimetallic NPs are shown in Fig. 7 (curves a-c). The Co NPs show no measurable features in the visible region, while the Au NPs display a surface plasmon resonance band at 520 nm. The surface plasmon resonance band of the Co–Au bimetallic NPs shows a red-shift and broadening of the peak compared to the Au NPs, which is commonly observed in other Au bimetallic systems.22,31
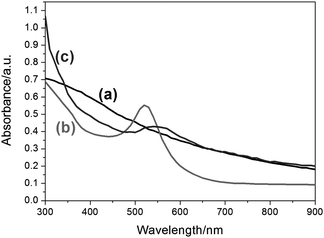 | ||
| Fig. 7 UV-visible spectra in toluene of (a) NaAOT coated Co NPs, (b) Au NPs and (c) Co–Au NPs. A sharp absorption peak can be observed for the Au NPs at 520 nm whereas for the Co–Au NPs, the absorption peak broadens, covering a range from 500 nm to 650 nm indicating the presence of Au. | ||
Similar shifts in the surface plasmon resonance band were observed in the spectra of the Fe3O4-Au NPs (Fig. 8, curves b and c), while the spectrum of Fe3O4 NPs was largely silent (Fig. 8 curve a), indicating the presence of Au in the former sample. No increase in the shift was observed for the thiolated DNA conjugated Fe3O4-Au NPs indicating that the NPs have not aggregated. The reduction in the intensity of curve c compared to curve b in Fig. 8, indicates that the concentration of the thiolated DNA conjugated Fe3O4-Au NPs was lower than the unconjugated Fe3O4-Au NPs.
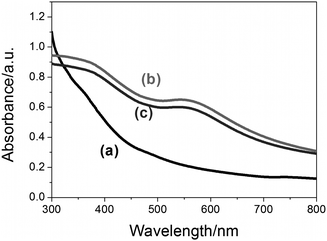 | ||
| Fig. 8 UV-visible spectra of (a) Fe3O4 NPs in hexane, (b) Fe3O4-Au NPs in water and (c) thiolated DNA conjugated to Fe3O4-Au NPs in water. The broad absorption peaks in the 500 nm to 650 nm range indicate the presence of Au in the samples. | ||
If the Fe3O4 core was homogeneously coated by the Au layer, the surface plasmon resonance (SPR) peak of Au would be shifted to longer wavelength compared to the SPR peak of spherical Au nanoparticles (∼520 nm) as calculated from the Mie theory (data not shown). In addition, the SPR band of the hollow Au shell would be broader than that of solid Au nanoparticles. Therefore, the broad SPR bands having a peak in the wavelength range from 550 nm to 600 nm (curves b and c in Fig. 8) suggest the existence of Au shells on the surfaces of Fe3O4 cores.
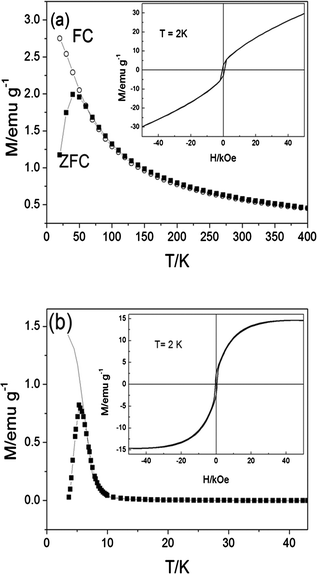 | ||
| Fig. 9 ZFC (symbols) and FC (line) magnetization curves of the NaAOT coated Co and Co–Au NPs as a function of temperature. The hysteresis curves measured at 2 K are shown in the insets. | ||
The ZFC and FC magnetization curves, as a function of temperature, of the Co and Co–Au NPs are shown in Fig. 9. There is a sharp peak in the ZFC curve and the splitting of the ZFC–FC occurs very close to the peak position suggesting a narrow size distribution for both the Co and Co–Au NPs,36 which is consistent with the TEM images in Fig. 1. From the peak position of the ZFC curves, the values of the blocking temperatures (TB) are obtained to be 40 K and 6 K for Co and Co–Au NPs, respectively. At an applied magnetic field of 100 Oe, the maximum magnetic susceptibility observed in the ZFC curve for Co NPs is 2.0 emu/g and Co–Au NPs 0.8 emu/g. The different magnetic properties observed between the Co and Co–Au NPs could be due to changes in the crystalline structure of the Co NPs as the gold atoms are added.31,37 It is also noted that Au metal itself does not contribute to the magnetism, but only adds mass to the NPs. It can be seen from the hysteresis curves measured at 2 K (insets of Fig. 9) that the Co and Co–Au NPs show behaviour like that of a conventional ferromagnet with observed coercivities of 650 Oe and 500 Oe, respectively. On the other hand, above the TB, no hysteresis was observed indicating that the particles are superparamagnetic (data not shown). The magnetization of the Co NPs measured at 2 K does not become saturated at the maximum applied magnetic field of 50 kOe which could be due to either the canting moments of the Co atoms at the surface38 or the presence of very small particles which are superparamagnetic even at very low temperature of 2 K.39 In addition, the NaAOT coated Co NPs are not very well protected from oxidation resulting in the formation of a thin antiferromagnetic CoO layer on the surface.40 This antiferromagnetic CoO layer also contributes to the observed unsaturated feature of the magnetization of the NaAOT coated Co NPs at high magnetic field. In contrast, the magnetization of the Co–Au NPs becomes saturated at approximately 15 emu/g, which may suggest that the presence of gold in the NPs offered a degree of protection from oxidation.
Fig. 10 shows the ZFC and FC magnetization curves, as a function of temperature, of the Fe3O4 and Fe3O4-Au NPs. The ZFC curves have peaks at the blocking temperature, TB = 15 K and TB = 29 K for the Fe3O4 and Fe3O4-Au NPs, respectively. In agreement with the narrow size distributions derived from TEM images in Fig. 3, we observe a sharp peak in the ZFC curve and the splitting of the ZFC and FC curves close to the peak position. Both, the Fe3O4 and Fe3O4-Au NPs display behavior like that of a conventional ferromagnet below TB and the coercivities derived from the hysteresis loops measured at 5 K are 90 Oe and 260 Oe, respectively. The increases in TB and coercivity observed for the Fe3O4-Au NPs could be associated with the increase in average diameter of the NPs, which leads to less effective coupling of the magnetic dipole moments of the cores.41 Above TB both types of NPs are superparamagnetic as indicated by the absence of measurable hysteresis. Similar magnetic properties were observed for both the Fe3O4-Au NPs and the thiolated DNA conjugated Fe3O4-Au NPs in aqueous solution (Fig. 10b and c). The saturation magnetisation of the Fe3O4 NPs was measured at 13 emu g−1, while the value for the Fe3O4-Au NPs was slightly lower at 12 emu g−1 due to the effect of the Au layer adding mass, but not to the magnetism of the NPs. This effect is further observed when the Fe3O4-Au NPs are conjugated with thiolated DNA (saturation magnetisation of these NPs was 11 emu g−1). This, along with the TEM and UV/vis spectra, indicates that the Fe3O4-Au NPs are stable in the presence of thiolated DNA. From dynamic light scattering data (Supporting information Figure 1S) it could be seen that the Fe3O4-Au NPs are monodisperse of 10 nm. This colloidal system is stable for at least one month when kept at room temperature.
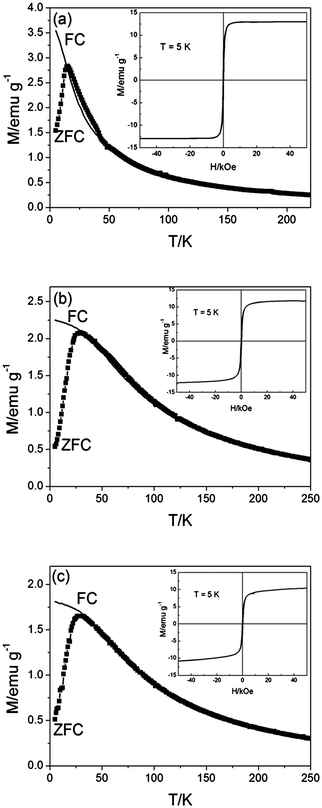 | ||
| Fig. 10 ZFC (symbols) and FC (line) magnetization curves of (a) Fe3O4 NPs in hexane, (b) Fe3O4-Au NPs in water and (c) thiolated DNA conjugated to Fe3O4-Au NPs in water, as a function of temperature. The hysteresis curves measured at 5 K are shown in the insets. | ||
Conclusion
The fabrication of core-shell magnetic NPs was attempted by the reduction of a gold salt on the surface of cobalt and iron oxide NPs. The gold layer can provide a versatile platform for the functionization of the NPs. The data acquired from the UV-visible and EDX spectra indicated that the Co–Au particles produced had a bimetallic structure. Further examination of the morphology by the use of HRTEM could not discern the presence of a core-shell structure. The absence of complete coverage of a Au shell on the surface of the NPs would also explain why attempts to stabilize these NPs using 11-mercaptoundecanoic acid resulted in agglomeration and precipitation of the NPs upon their transfer to aqueous solution. On the other hand, the larger size of the Fe3O4-Au NPs compared to the Fe3O4 seeds suggested that a Au layer had successfully been added to the latter. The presence of a Fe3O4-Au core-shell structure was further supported by HRTEM, where two distinct layers with different lattice constants were observed. The presence of a Au layer enabled the functionalization of the magnetic NPs with thiolated DNA, which remained stable in aqueous solution. This interaction between the Au and thiol groups could be exploited further to conjugate, through specific and covalent chemistry, a wide variety of molecules to the NPs, making them extremely versatile for a large range of applications.Acknowledgements
Nguyen TK Thanh thanks The Royal Society for her University Research Fellowship. This work was in part funded by the Engineering and Physical Sciences Research Council, UK (EPSRC). Ian Prior and Simon Romani are thanked for provision of the TEM and EDX facilities, respectively. Joshua Owen is thanked for technical support. Christoph Wälti acknowledges the support of the RCUK's Basic Technology programme.Notes and References
- C. C. Berry and A. S. G. Curtis, J. Phys. D: Appl. Phys., 2003, 36, R198–R206 CrossRef CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS.
- N. T. K. Thanh, I. Robinson and L. D. Tung, Dekker Encycloped. Nanosci. Nanotechnol., 2007, 1, 1–10 Search PubMed.
- L. M. Parkes, R. Hodgson, L. T. Lu, L. D. Tung, I. Robinson, D. G. Fernig and N. T. K. Thanh, Contrast Media Mol. Imaging, 2008, 3, 150–156 Search PubMed.
- Q. A. Pankhurst, N. K. T. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001–224015 CrossRef.
- M. Giersig and M. Hilgendorff, J. Phys. D: Appl. Phys., 1999, 32, L111–L113 CAS.
- C. B. Murray, S. H. Sun, W. Gaschler, H. Doyle, T. A. Betley and C. R. Kagan, IBM J. Res. Dev., 2001, 45, 47–56 CrossRef CAS.
- I. Robinson, M. Volk, L. D. Tung, G. Caruntu, N. Kay and N. T. K. Thanh, J. Phys. Chem. C, 2009, 113, 9497–9501 CrossRef CAS.
- I. Robinson, S. Zacchini, L. D. Tung, S. Maenosono and N. T. K. Thanh, Chem. Mater., 2009, 21, 3021–3026 CrossRef CAS.
- L. E. Euliss, S. G. Grancharov, S. O'Brien, T. J. Deming, G. D. Stucky, C. B. Murray and G. A. Held, Nano Lett., 2003, 3, 1489–1493 CrossRef CAS.
- N. T. K. Thanh, V. F. Puntes, L. D. Tung and D. G. Fernig, J. Phys. Conf. Ser., 2005, 17, 70–76 CrossRef CAS.
- R. Narain, M. Gonzales, A. S. Hoffman, P. S. Stayton and K. M. Krishnan, Langmuir, 2007, 23, 6299–6304 CrossRef CAS.
- I. Robinson, C. Alexander, L. T. Lu, L. D. Tung, D. G. Fernig and N. T. K. Thanh, Chem. Commun., 2007, 4602–4604 RSC.
- T. R. Zhang, J. P. Ge, Y. P. Hu and Y. D. Yin, Nano Lett., 2007, 7, 3203–3207 CrossRef CAS.
- L. T. Lu, L. D. Tung, I. Robinson, D. Ung, B. Tan, J. Long, A. I. Cooper, D. G. Fernig and N. T. K. Thanh, J. Mater. Chem., 2008, 18, 2453–2458 RSC.
- I. Robinson, C. Alexander, L. D. Tung, D. G. Fernig and N. T. K. Thanh, J. Magn. Magn. Mater., 2009, 321, 1421–1423 CrossRef CAS.
- Y. W. Jun, Y. M. Huh, J. S. Choi, J. H. Lee, H. T. Song, S. Kim, S. Yoon, K. S. Kim, J. S. Shin, J. S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS.
- S. Y. Lee and M. T. Harris, J. Colloid Interface Sci., 2006, 293, 401–408 CrossRef CAS.
- R. De Palma, S. Peeters, M. J. Van Bael, H. Van den Rul, K. Bonroy, W. Laureyn, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef CAS.
- L. Y. Wang, J. Luo, Q. Fan, M. Suzuki, I. S. Suzuki, M. H. Engelhard, Y. H. Lin, N. Kim, J. Q. Wang and C. J. Zhong, J. Phys. Chem. B, 2005, 109, 21593–21601 CrossRef CAS.
- W. R. Lee, M. G. Kim, J. R. Choi, J. I. Park, S. J. Ko, S. J. Oh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 16090–16097 CrossRef CAS.
- J. L. Lyon, D. A. Fleming, M. B. Stone, P. Schiffer and M. E. Williams, Nano Lett., 2004, 4, 719–723 CrossRef CAS.
- V. Salgueirino-Maceira and M. Correa-Duarte, Adv. Mater., 2007, 19, 4131–4144 CrossRef.
- Z. C. Xu, Y. L. Hou and S. H. Sun, J. Am. Chem. Soc., 2007, 129, 8698 CrossRef CAS.
- M. Pita, J. Abad and C. Vaz-Dominguez, J. Colloid Interface Sci., 2008, 321, 484–492 CrossRef CAS.
- Z. H. Lu, M. D. Prouty, Z. H. Guo, V. O. Golub, C. S. S. R. Kumar and Y. M. Lvov, Langmuir, 2005, 21, 2042–2050 CrossRef CAS.
- Z. H. Ban, Y. A. Barnakov, V. O. Golub and C. J. O'Connor, J. Mater. Chem., 2005, 15, 4660–4662 RSC.
- Q. X. Liu, Z. H. Xu, J. A. Finch and R. Egerton, Chem. Mater., 1998, 10, 3936–3940 CrossRef CAS.
- S. Mandal and K. M. Krishnan, J. Mater. Chem., 2007, 17, 372–376 RSC.
- J. A. Barnard, M. R. Parker, D. Seale and J. Yang, IEEE Trans. Magn., 1994, 29, 2711–2713.
- G. J. Cheng and A. R. Hight Walker, J. Magn. Magn. Mater., 2007, 311, 31–35 CrossRef CAS.
- X. W. Teng, D. Black, N. J. Watkins, Y. L. Gao and H. Yang, Nano Lett., 2003, 3, 261–264 CrossRef CAS.
- M. Mikhaylova, D. K. Kim, N. Bobrysheva, M. Osmolowsky, V. Semenov, T. Tsakalakos and M. Muhammed, Langmuir, 2004, 20, 2472–2477 CrossRef CAS.
- M. Mandal, S. Kundu, S. K. Ghosh, S. Panigrahi, T. K. Sau, S. M. Yusuf and T. Pal, J. Colloid Interface Sci., 2005, 286, 187–194 CrossRef CAS.
- N. K. T. Thanh and L. A. W. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- J. S. Micha, B. Dienyb, J. R. Régnard, J. F. Jacquot and J. Sortb, J. Magn. Magn. Mater., 2004, S1, 272–276.
- B. Y. Tsaur, S. S. Lau and J. W. Mayer, Phil. Mag. B-Phys. Condens. Matter Statistical Mechanics Electronic Optical Magn. Prop., 1981, 44, 95–108 Search PubMed.
- M. P. Morales, C. J. Serna, F. Bødker and S. Mørup, J. Phys.: Condens. Matter, 1997, 9, 5461 CrossRef CAS.
- G. Dietz and H. D. Schneider, J. Phys.: Condens. Matter, 1990, 2, 2169 CrossRef CAS.
- A. N. Dobrynin, D. N. Ievlev, K. Temst, P. Lievensa, J. Margueritat, J. Gonzalo, C. N. Afonso, S. Q. Zhou, A. Vantomme, E. Piscopiello and G. V. Tendeloo, Appl. Phys. Lett., 2005, 87, 012501–012503 CrossRef.
- A. K. Boal, B. L. Frankamp, O. Uzun, M. T. Tuominen and V. M. Rotello, Chem. Mater., 2004, 16, 3252–3256 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Dynamic light scattering results. See DOI: 10.1039/c0nr00621a |
| This journal is © The Royal Society of Chemistry 2010 |