Linear release nanoparticle devices for advanced targeted cancer therapies with increased efficacy†
Alice E.
van der Ende
a,
Vasanth
Sathiyakumar
a,
Roberto
Diaz‡
b,
Dennis E.
Hallahan§
b and
Eva
Harth
*a
aDepartment of Chemistry, Vanderbilt University, 7619 Stevenson Center, Nashville, Tennessee 37235, USA. E-mail: eva.harth@vanderbilt.edu; Fax: +01 615 343 1234; Tel: +01 615343 3405
bDepartment of Radiation Oncology, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA
First published on 4th January 2010
Abstract
The cross-linked supramolecular structure of prepared polyester based nanoparticles enable increased and efficient small molecule drug loading after nanoparticle formation and post-modification with targeting peptides via thiol-ene ‘click’ chemistry. It could be demonstrated that the drug loading does not influence the structural integrity of the particle and its diameter was not significantly changed. A prolonged linear release profile of the drug without the typical ‘burst effect’ was observed in emulsified particles and mirrored the linear degradation profile of the investigated particles, which are critical properties for controlled and predictable pharmacokinetics in cancer therapies. Furthermore, the final peptide-targeted and drug-loaded particles were found to be readily dispersed in buffer or water, and with the confirmed biocompatibility, these novel and adjustable drug delivery systems are promising vectors for the treatment of various cancers.
Introduction
Traditional polyester nanoparticle delivery systems are typically self-assembled from linear polyesters chains driven by the polarity of the solvent, emulsion composition and addition techniques.1–3 These procedures predetermine the drug loading during nanoparticle formation and limit post-modification chemistries in organic and aqueous solutions.4 Furthermore, the result of this self-assembly process is mirrored in the morphology and degradation properties of the release systems. It has been recognized that the degradation behavior of the nanoparticles and release profile of the entrapped drug molecules are the key factors to establish predictable pharmacokinetic profiles5,6 in effective multidrug cancer therapies. So far, release kinetics are challenged by a rapid release of the drug molecules in the first 24–48 h followed by a slower release, referred to as a ‘burst-effect’.2,7 These release profiles prevent the establishment of reliable dosages and contribute to developing multidrug resistance, often times the result of non-optimized drug concentrations at tumor sites.In this work, we present the synthesis and evaluation of actively targeted drug delivery carriers that are shown to be unique in their capability to entrap high concentrations of hydrophobic therapeutics and maintain a linear release profile which can be tuned to the demands of the tumor type as a result of the adjustable supramolecular architecture accomplished through an intermolecular cross-linking technique.
The novel method to prepare polyester particles relies on a controlled cross-linking mechanism of linear polyester precursors that contain pendant functional groups as one of the critical cross-linking units with a difunctionalized linker that acts as the second cross-linking partner.8,9 To achieve control over a series of different nanoparticle size dimensions, the amount of the difunctionalized linker is critical and is added in a series of varying equivalencies to the pendant functionalities of the linear polyester precursor. In this fashion, nanoparticles can be produced, depending on the linker amount present in the reaction, with unique sizes and standard deviations of only 10%, as we have reported recently.9 Furthermore, we could show that these ‘nano-networks’, depending on their nanoparticle size and cross-linking density, influence their crystallinity, but all particles are amorphous at the intended temperature of use (37 °C).
Discussion
To determine if the amorphous properties of poly(valerolactone-epoxyvalerolactone), poly(vl-evl)8 particles have a positve effect on the degradation behavior, we completed a series of degradation studies in buffer at pH 7.4 at 37 °C, investigating particles from a completed series of linear precursors and increasing amounts of difunctionalized cross-linkers (Fig. 1). The degradation of the particles was monitored by the change of the absolute molecular weight, as determined through static light scattering (SLS). We observed linear degradation profiles for all particles investigated, with the highest loss of molecular weight for the 725 nm nanoparticle with 17.5% of its total molecular mass remaining after 10 days. Smaller particles with a slightly higher degree of crystallinity of 20.6% were degraded to 26% of the original molecular weight. The observed linear degradation kinetics are one critical parameter that will determine the quality of the developed particles towards applications as controlled release systems.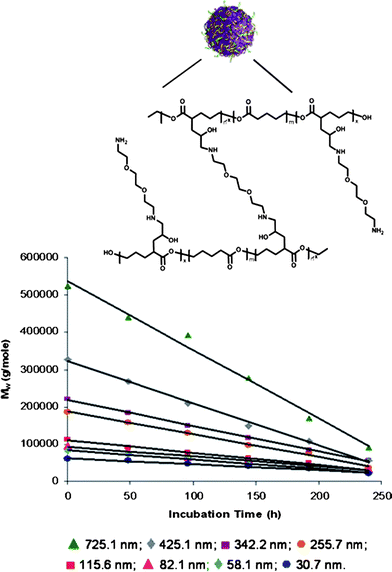 | ||
| Fig. 1 Hydrolytic biodegradation studies of (▲) 725.1 ± 94.3 nm poly(vl-evl) nanoparticles to (♦) 30.71 ± 2.21 nm AB nanoparticles. All particles are non-emulsified. | ||
As a next step, it will be necessary to investigate the capacity to encapsulate small molecule drugs, such as paclitaxel (taxol). Traditional polyester particles, produced with salting-out or nanoprecipitation methods, typically do not exceed a drug loading over 5% that is facilitated during nanoparticle formation. However, the developed nanoparticles consist of cross-linked supramolecular structures that are readily soluble in organic solvents without affecting the 3-D architecture. This property gives us the opportunity to load the particles after formation by dissolving the particles in dimethyl sulfoxide (DMSO) together with cancer therapeutics, such as paclitaxel (taxol), and precipitating into water. The discussed experiments to determine the drug loading capacity were performed with particles of 53 nm in diameter from linear precursors, poly(valerolactone-epoxyvalerolactone-allylvalerolactone-oxepanedione), poly(vl-evl-avl-opd),8 containing 11% epoxide and cross-linked with 2 equivalents of diamines per epoxide (Fig. 3). In view of future in vivo experiments, we chose to optimize the encapsulation method to also increase the homogenity of the particle dispersion in water for a practical adminstration of the drug loaded particles by injection. Here, we utilized an emulsification process using vitamin E TPGS (D-α-tocopherol polyethylene glycol 1000 succinate),10 that achieves a homogenous dispersion of the loaded or un-loaded particles in water or buffer. The resulting particles are analyzed by UV-Vis with a NanoDrop™ Spectrophotometer at 254 nm, and along with a calibration curve, the drug loading of paclitaxel was found to be 15.7% for an aimed 20% drug load and 11.3% for a 15% drug load, respectively (see ESI†). We can conclude that with this process, it is not only possible to load therapeutic drug molecules to a higher degree into prepared nanoparticles, but we have found also a method for the solubilization of hydrophobic cancer therapeutics in aqueous solutions. As a result, it can be anticipated that side effects known to be caused by adjuvant agents, such as Cremophor® EL (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ethanol–polyoxyethylated castor oil) to solubilize hydrophobic drug molecules for intravenous injections, can be avoided.
:50 ethanol–polyoxyethylated castor oil) to solubilize hydrophobic drug molecules for intravenous injections, can be avoided.
To ensure that no cellular toxicity is caused by the vitamin E TPGS formulated particles prior to drug loading, the cell viability was assessed by utilizing a MTT assay2 (Fig. 2). The cellular toxicity was determined by incubating HeLa cells with varying concentrations of particles in triplicate ranging from 5 mg ml−1 to 0.001 mg ml−1. Following 24 h of incubation with particles, cell viability was assessed. As seen in Fig. 2, the nanoparticles did not cause significant cytotoxicity against the HeLa cell line. The experimental TC50 value for the formulated particles was found to be 1.0 mg mL−1.
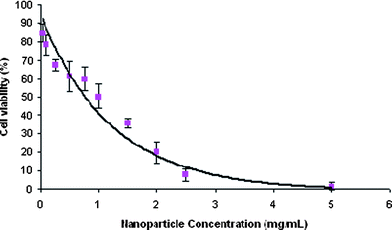 | ||
| Fig. 2 Cytotoxicity of vitamin E TPGS formulated nanoparticles on HeLa cells after 24 h incubation using the MTT assay. Fitted curve shows cell viability of the HeLa cell line. | ||
Moreover, we found that the emulsification had a profound effect on the degradation profile and found to correlate with the in vitro release studies. Over the period of 16 days, the particles experienced, a low controlled degradation, as seen by the linear degradation, finishing with 70% of its original molecular weight remaining (Fig. 3). The slower degradation rate can be attributed to the well-defined structure of the nanoparticle and the vitamin E TPGS that might remain present at the surface to stabilize the particles. Consequently, this gradual constant degradation profile of the particles is a desirable feature as it could translate into the controlled and sustained release of therapeutics.
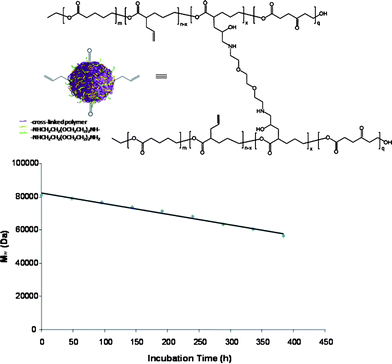 | ||
| Fig. 3 In vitro degradation profile of vitamin E TPGS formulated poly(vl-evl-avl-opd) nanoparticles of 53 nm in DPBS at pH 7.4 and 37 °C over a period of 384 h (16 days). | ||
After establishing procedures for the loading and solublization of drug molecules in the particles, we wanted to evaluate the drug release profile in vitro. The paclitaxel release kinetics from vitamin E TPGS formulated nanoparticles were assessed by monitoring the cumulative release of taxol at 37 °C in DPBS at pH 7.4. At particular time intervals, the samples were centrifuged and the supernatant was taken for analysis of paclitaxel concentration by NanoDrop™ spectrophotometry (254 nm). Fig. 4 depicts the cumulative release of paclitaxel from the particles. The profile shows a collective release of 4.4% and 7.4% taxol in the first 2 and 6 h respectively, followed by a slow and sustained release over 60 days, which again confirmed the efficient encapsulation of paclitaxel within the cross-linked nanoparticles. The initial instant release of paclitaxel in the first several hours may be due to the dissolution or diffusion of the drug that was absorbed onto the nanoparticle surface, while the linear slow continuous release may be attributed to the diffusion of the drug encapsulated in the nanoparticle during degradation. Traditional poly(lactic-co-glycolic acid) (PLGA) nanoparticles, however, experience an erratic nonlinear drug release, that includes a ‘burst-effect’ in which about 40% of taxol is released in the first day, followed by a fast release of about 10–30% in the next 2–5 days and then finally a slow release till no paxlitaxel (taxol) remains.11 The comparison of these release studies to other traditional polyester based particles is given in the ESI (Fig. S4a).† We have to point out that the release kinetics can be adjusted to faster or slower release governed by the density of cross-linking and the particle size.
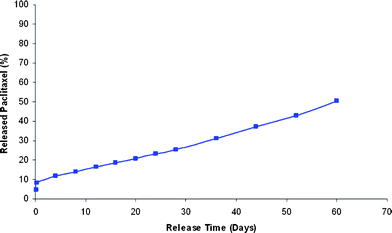 | ||
| Fig. 4 In vitro release profile of paclitaxel from particles loaded with 11.3% paclitaxel prepared with the emulsification process. The drug release was performed in DPBS at pH 7.4 and 37 °C for 60 days. The cumulative release profile shows a desirable controlled and sustained release of paclitaxel from the nanoparticles. | ||
In view of future in vivo studies in which the particle sizes play an important role in the interaction with the tumor vasculature, we wanted to evaluate the influence of formulation and encapsulation of small molecule drugs on the diameter of the nanoparticles. Interestingly, we found that based on the 3-D cross-linked network structure, the size dimension slightly changes from 53 nm to 57 nm and indicates the conformity of the 3-D network structure upon encapsulation, as seen by transmission electron microscopy (TEM), (Fig. 5), with 2–8 times more drug incorporated compared to traditional polyester nanoparticle systems.
 | ||
| Fig. 5 Transmission electron microscopy (TEM) images of (A) nanoparticles without taxol with a size of 53 nm and (B) nanoparticles encapsulated with 11.3% taxol with a size dimension of 57 nm. | ||
Encouraged by the linear release kinetic profiles and structural integrity of the investigated particles, we sought to advance theses controlled release systems into targeted nanoparticles for an evaluation of these properties in vivo. As previously reported, the developed nanoparticle synthesis pathway allows for the introduction of functional groups, such as alkyne, allyl or keto functionalities, that are not affected by the cross-linking reactions and nanoparticle formation.8 In particular, we have optimized thiol-ene ‘click’ reactions that allow for the conjugation of peptides with integrated cysteines added to the sequence near the N-terminus. The mild reaction conditions do not require the addition of radical starters and use slightly elevated temperatures of 37 °C.9 To synthesize the drug delivery system we first prepared the linear poly(vl-evl-avl-opd) precursor which was cross-linked with 2 equivalents of diamines per epoxide to form a nanoparticle of 53 nm in size.9 The remaining allyl groups were then functionalized with peptides to target radiated12 and non-radiated13 tumor vasculature, such as the reported peptides with recognition units HVGGSSV14 and cRGD,9 respectively (Fig. 6). The bioconjugates were analyzed via NMR, DLS and SLS and were then loaded with paclitaxel and formulated with vitamin E. Using UV-Vis, the loading capacity was found to be 11%, aiming for a 15% drug load. The in vivo investigation of the targeted particles in tumor suppression studies will be published in due course.
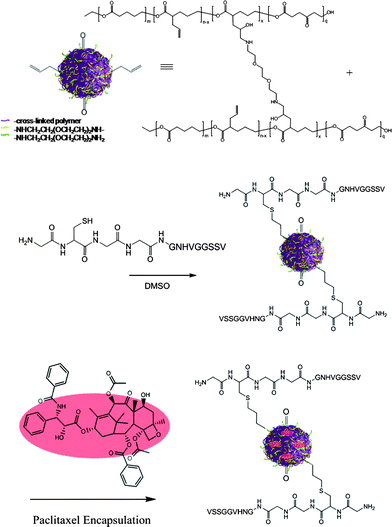 | ||
| Fig. 6 Synthesis of a targeted nanoparticle drug delivery system involving thiol-ene ‘click’ chemistries and drug loading via developed emulsification process after post-modification. | ||
Conclusions
In summary, we could demonstrate that the previously developed intermolecular cross-linking technique leading to supramolecular 3D nano-networks provided the basis for the preparation of targeted and sustained delivery systems. It was found that small drug molecules can be incorporated after nanoparticle formation, with and without targeting units attached, and a higher efficacy in drug loading was achieved than reported for traditional polyester particle systems. Furthermore, an applied emulsification approach during drug loading could prolong the linear degradation rate and resulted in the water-dispersion of the final targeted drug delivery systems with paclitaxel incorporated. The particle sizes did not change significantly upon loading, which can contribute to the stable, cross-linked supramolecular structure of the 3-D nano-networks. Based on the versatility of nanoparticle formation and adjustment of cross-linking density to achieve different degradation profiles, the technique provides a means to adjust release profiles of the drug delivery systems to the physiological demands in vivo. The observed linear release kinetics of paclitaxel without a ‘burst-effect’ are another indication that the developed techniques for nanoparticle formation, encapsulation and post-modification will provide effective targeted delivery systems with predictable pharmacokinetic profiles that are crucial to the development of any meaningful clinical protocols.Acknowledgements
E.H. gratefully acknowledges Vanderbilt University with a startup fund and finacial support from the NSF under Award CHE-0645737. A. van der Ende acknowledges Vanderbilt University for a Warren Fellowship.Notes and references
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS.
- J. M. Chan, L. F. Zhang, K. P. Yuet, G. Liao, J. W. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 CrossRef CAS.
- M. N. V. R. Kumar, U. Bakowsky and C. M. Lehr, Biomaterials, 2004, 25, 1771–1777 CrossRef CAS.
- J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Biomaterials, 2007, 28, 869–876 CrossRef CAS.
- A. S. Hasan, M. Socha, A. Lamprecht, F. El Ghazouani, A. Sapin, A. Hoffman, P. Maincent and N. Ubrich, Int. J. Pharm., 2007, 344, 53–61 CrossRef CAS.
- A. S. Hassan, A. Sapin, A. Lamprecht, E. Emond, F. El Ghazouni and P. Mainchent, Eur. J. Pharm. Biopharm., 2009 DOI:10.1016/j.ejpb.2009.07009.
- S. S. Feng, L. Y. Zhao, Z. P. Zhang, G. Bhakta, K. Y. Win, Y. C. Dong and S. Chien, Chem. Eng. Sci., 2007, 62, 6641–6648 CrossRef CAS.
- A. E. van der Ende, E. J. Kravitz and E. Harth, J. Am. Chem. Soc., 2008, 130, 8706–8713 CrossRef CAS.
- A. van der Ende, T. Croce, S. Hamilton, V. Sathiyakumar and E. Harth, Soft Matter, 2009, 5, 1417–1425 RSC.
- L. Mu and S. S. Feng, J. Controlled Release, 2002, 80, 129–144 CrossRef CAS.
- L. Vicari, T. Musumeci, I. Giannone, L. Adamo, C. Conticello, R. De Maria, R. Pignatello, G. Puglisi and M. Gulisano, BMC Cancer, 2008, 8, 212 CrossRef.
- R. Diaz, R. J. Passarella and D. E. Hallahan, Expert Rev. Anticancer Ther., 2008, 8, 1787–1796 Search PubMed.
- E. A. Murphy, B. K. Majeti, L. A. Barnes, M. Makale, S. M. Weis, K. Lutu-Fuga, W. Wrasidlo and D. A. Cheresh, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9343–9348 CrossRef CAS.
- Z. Han, A. Fu, H. Wang, R. Diaz, L. Geng, H. Onishko and D. E. Hallahan, Nat. Med., 2008, 14, 343–349 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Nanoparticle synthesis and characterization, procedures and analytical methods. See DOI: 10.1039/b9py00272c |
| ‡ Present address: Department of Radiation Oncology, Emory University School of Medicine, 1365 Clifton Road, NE, Suite A1300, Atlanta, Georgia, 30322, USA. |
| § Present address: Department of Radiation Oncology, Mallinckrodt Institute of Radiology; and Siteman Cancer Center, Washington University School of Medicine, St Louis, Missouri, 63110, USA. |
| This journal is © The Royal Society of Chemistry 2010 |