DOI:
10.1039/B9PY00223E
(Paper)
Polym. Chem., 2010,
1, 82-92
Hyperbranched 5,6-glucan as reducing sugar ball†
Received
23rd August 2009
, Accepted 2nd November 2009
First published on
21st December 2009
Abstract
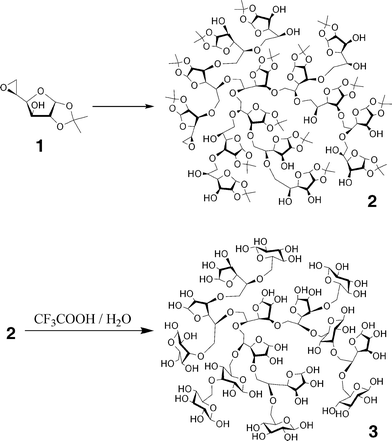
The ring-opening polymerization of 5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose (1) as a latent cyclic AB2-type monomer was carried out using potassium tert-butoxide (t-BuOK) or boron trifluoride diethyletherate (BF3·OEt2) as an initiator in order to synthesize a novel hyperbranched glycopolymer. The anionic and cationic polymerizations proceeded via the proton-transfer reaction mechanism to produce the hyperbranched poly(5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose) (2). In particular, the cationic polymerization with the slow-monomer-addition strategy is a facile method leading to the hyperbranched glycopolymers with high molecular weights and highly branched structures. The weight-average molecular weight (Mw,SEC-MALLS) values of 2 measured by multi-angle laser light scattering (MALLS) varied in the range from 7400 to 122![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, which were significantly higher than the weight-average molecular weight (Mw,SEC) values determined by size exclusion chromatography (SEC). The intrinsic viscosities ([η]) of these polymers were very low in the range of 3.3–4.6 mL g−1 and the Mark–Houwink–Sakurada exponents, α, were calculated to be 0.08–0.27. These results of the MALLS, SEC, and viscosity measurements suggested that these polymers exist in a compact spherical conformation in solution because of their highly branched structure. The synthesis of the hyperbranched 5,6-glucan (3) by hydrolysis of polymer 2 was also demonstrated. Polymer 3 is a novel water-soluble hyperbranched glycopolymer arranged with numerous reducing D-glucose units on the peripheries of the polymer, and has a higher reducing ability than D-glucose because of the glyco-cluster effect or the multivalent effect of the reducing D-glucose units. Therefore, polymer 3 should be called a “reducing sugar ball”.
400, which were significantly higher than the weight-average molecular weight (Mw,SEC) values determined by size exclusion chromatography (SEC). The intrinsic viscosities ([η]) of these polymers were very low in the range of 3.3–4.6 mL g−1 and the Mark–Houwink–Sakurada exponents, α, were calculated to be 0.08–0.27. These results of the MALLS, SEC, and viscosity measurements suggested that these polymers exist in a compact spherical conformation in solution because of their highly branched structure. The synthesis of the hyperbranched 5,6-glucan (3) by hydrolysis of polymer 2 was also demonstrated. Polymer 3 is a novel water-soluble hyperbranched glycopolymer arranged with numerous reducing D-glucose units on the peripheries of the polymer, and has a higher reducing ability than D-glucose because of the glyco-cluster effect or the multivalent effect of the reducing D-glucose units. Therefore, polymer 3 should be called a “reducing sugar ball”.
Introduction
The spherical architectures of highly branched macromolecules, such as dendrimers, star-shaped polymers, and hyperbranched polymers, have attracted much attention from the viewpoint of nanotechnology, because their numerous terminal units can be converted into various functional groups leading to novel nanomaterials.1–32 Thus, various types of dendrimers, star polymers, and hyperbranched polymers have been synthesized and their properties were compared to the linear analogues.23,33–46
Spherical macromolecular architectures with their surfaces covered with sugars are of increasing interest due to their unique structures along with specific properties, such as the glycol-cluster or multivalent effect on the carbohydrate-protein interactions. For a spherical polymer with many terminal sugar residues, there are a few types of glycodendrimers, e.g., dendrimers prepared from the reaction of the dendrimer surface with mono- and disaccharides (sugar ball),47–59 and dendrimers prepared through the convergent method using semi-dendritic branched oligosaccharides.60–64 Although the synthesis of such glycodendrimer types was accomplished, dendrimers essentially consisting of sugar units, i.e., dendritic carbohydrate polymers, have not been prepared until now due to the complicated and time-consuming synthetic process involving numerous protection, deprotection, and purification procedures. Star-shaped glycopolymers were also prepared using the “core-first” method based on the RAFT polymerization65,66 and the “arm-first” method based on the TEMPO-mediated radical polymerization.67–70 Hyperbranched glycopolymers from the polymerization of ABm-type monomers can be prepared more easily than glycodendrimers and star-shaped glycopolymers.71–76 Recently, we proposed that the ring-opening multibranching polymerization of anhydro and dianhydro sugars as latent ABm-type monomers should be a convenient method of synthesizing hyperbranched glycopolymers with a novel spherical macromolecular architecture,77–83 for example, the hyperbranched polysaccharides from the cationic polymerization of 1,6-anhydro-β-D-hexopyranose, the hyperbranched poly(2,5-anhydro-D-glucitol) from 1,2:5,6-dianhydro-D-mannitol, and the hyperbranched polytetritols from 1,4-anhydrotetritol and 2,3-anhydrotetritol. For our strategy for synthesising hyperbranched glycopolymers, it is important to elucidate the effect of the structure of the sugar monomers on the ring-opening multibranching polymerization tendency and to clarify the relationship between the structure and properties of the hyperbranched glycopolymers.
In this article, we report the ring-opening multibranching polymerization of 5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose (1) as a latent cyclic AB2-type monomer using anionic or cationic initiators, as shown in Fig. 1, leading to the hyperbranched poly(5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose) (2). In fact, Uryu et al. previously reported pioneering research on the ring-opening polymerization of 1,84,85 although the polymer structure and the solution properties were insufficiently characterized. Therefore, the characteristics of the polymerization, the polymer structure, and the solution characteristics were discussed by the NMR measurements and the size exclusion chromatography (SEC) with a multi-angle laser light scattering (MALLS) detector and a viscosity detector. In addition, the synthesis of a hyperbranched 5,6-glucan (3) by hydrolysis of polymer 2 was demonstrated. Since polymer 3 is a novel water-soluble hyperbranched glycopolymer arranged with numerous reducing D-glucose units on the peripheries of the polymer, the reduction property of 3 is discussed from the viewpoint of the physical property as a “reducing sugar ball”.
Experimental
Materials
5,6-Anhydro-1,2-O-isopropylidene-α-D-glucofuranose (1) was synthesized from 1,2-O-isopropylidene-α-D-glucofuranose using a procedure similar to that reported by Yus et al.861 was purified by column chromatography on silica gel with n-hexane/ethyl acetate (3/2, v/v, Rf 0.15) and dried in a vacuum oven at 50 °C for 2 days. 1H NMR (CDCl3):δ (ppm) 1.32 and 1.48 (s, 6H, 2CH3), 2.88 and 3.00 (m, 2H, 2H-6), 3.11 (m, 1H, OH), 3.43 (m, 1H, H-5), 4.07 (m, 1H, H-4), 4.26 (m, 1H, H-3), 4.52 (m, 1H, H-2), 5.99 (d, 1H, H-1). 13C NMR (CDCl3):δ (ppm) 26.1 and 26.7 (2C, 2CH3), 45.9 (C-6), 50.2 (C-5), 75.1 (C-3), 79.2 (C-4), 85.0 (C-2), 104.9 (C-1), 111.8 (C). Potassium tert-butoxide (t-BuOK)(1.0 mol L−1 in THF) was obtained from the Sigma-Aldrich Co., and used as received. Boron trifluoride diethyletherate (BF3·OEt2) was purchased from Kanto Chemical Co., Ltd (Tokyo, Japan) and distilled over CaH2 under reduced pressure. Silica gel 60N (Kanto Chemical Co., Ltd; spherical shape; particle size, 40–50 μm; neutral) was used for the column chromatography. Methyl iodide (>99.5%), silver oxide (>99.0%), an ammonia aqueous solution (28.0–30.0%), tetrachloroauric (III) acid (HAuCl4) tetrahydrate (>99.0%), trifluoroacetic acid (>99.0%), dry dichloromethane (dry CH2Cl2) (>99.5%; water content, <0.001%), dry tetrahydrofuran (THF) (>99.5%; water content, <0.001%), dry acetonitrile (CH3CN) (>99.5%; water content, <0.001%), methanol (>99.5%), acetone (>99.0%), n-hexane (>95.0%), ethyl acetate (>99.5%), dimethylformamide (DMF) (>99.5%), and chloroform (CHCl3) (>99.0%) were obtained from Kanto Chemical Co., Ltd, and used without further purification.
Measurements
The spectra of 1H (400 MHz), 13C NMR (100 MHz), distortionless enhancement by polarization transfer (DEPT), and 13C-1H correlation spectroscopy (COSY) were recorded using a JEOL JNM-A400II instrument. The molecular weight values of polymers 2, 4 and 6 were determined by size exclusion chromatography (SEC) in THF (1.0 mL min−1) at 40 °C using an Agilent 1100 series instrumentation equipped with two Shodex KF-804L columns (linear, 8.0 mm × 300 mm; exclusion limit, 4 × 105; bead size, 7 μm), a DAWN 8 multi-angle laser light scattering (MALLS) detector (Wyatt Technology, Santa Barbara, CA), a Viscostar viscosity detector (Wyatt Technology), and an Optilab rEX refractive index detector (Wyatt Technology). The absolute molecular weights (Mw,SEC-MALLS), polydispersity (Mw,SEC-MALLS/Mn,SEC-MALLS), intrinsic viscosity ([η]), refractive index change (dn·dc−1), and Mark–Houwink–Sakurada constants, α and K, were estimated by ASTRA 5.1.6.0 software (Wyatt Technology). The relative molecular weight (Mw,SEC) and the polydispersity (Mw,SEC/Mn,SEC) in THF were calculated on the basis of a polystyrene calibration. The molecular weight values of polymer 3 were determined by size exclusion chromatography (SEC) in a 0.2 mol L−1 NaNO3 aqueous solution (1.0 mL min−1) at 40 °C using an Agilent 1100 series instrumentation equipped with two Tosoh TSKgel GMPWXL columns (linear, 7.8 mm × 300 mm; exclusion limit, 5 × 107; bead size, 13 μm), a DAWN 8 multi-angle laser light scattering (MALLS) detector (Wyatt Technology), a Viscostar viscosity detector (Wyatt Technology), and an Optilab rEX refractive index detector (Wyatt Technology). The Mw,SEC-MALLS, Mw,SEC-MALLS/Mn,SEC-MALLS, [η], dn·dc−1, and Mark–Houwink–Sakurada constants, α and K, were estimated by ASTRA 5.1.6.0 software (Wyatt Technology). Preparative SEC for the chloroform-soluble polymers was performed in chloroform (3.5 mL min−1) at 23 °C using a JAI LC-9201 (Japan Analytical Industry Co., Ltd, Japan) equipped with a JAI JAIGEL-3H column (20 mm × 600 mm; exclusion limit, 7 × 104) and a JAI RI-50s refractive index detector. The preparative SEC for the water-soluble polymers was performed in water (14 mL min−1) at 23 °C using a JAI LC-928 (Japan Analytical Industry Co., Ltd, Japan) equipped with a JAI JAIGEL-W253-40 column (40 mm × 500 mm; exclusion limit, 5 × 104) and a JAI RI-50s refractive index detector. The elemental analysis was performed using a MICRO CORDER JM10 and Dionex DX-500 (Instrumental Analysis Division, Management Center, Creative Research Instaction Sousei, Hokkaido University). The measurement of the optical rotations was performed in CHCl3 or H2O using a Jasco DIP-1000 digital polarimeter (λ = 589 nm). The FI-MASS spectroscopy measurement was performed using a JEOL JMS-SX102A mass spectrometer (GC-MS and NMR Laboratory, Graduate School of Agriculture, Hokkaido University). The UV-vis spectra were measured at 70 °C with a 10 mm path length using a JASCO V-550 spectrometer with a deuterium lamp as the light source for the UV range (190–350 nm) and a halogen lamp for the visible range (330–900 nm). The dynamic light scattering (DLS) measurement of the obtained Au particles was performed at 25 °C using an Otsuka Electronics FDLS-3000 light scattering spectrophotometer equipped with a solid state laser (λ = 532 nm, scattering angle = 90°). Before the measurement, the samples were filtered using 0.45 μm PTFE membrane filters to eliminate any dust particles. The data analysis was carried out using the histogram methods including the Marquadt analysis.
All the anionic polymerizations of 1 were carried out using a storage flask with a needle valve under an argon atmosphere. A typical polymerization procedure (entry 2 in Table 1) is as follows: 1.0 mol L−1t-BuOK in a THF solution (0.50 mL, 0.50 mmol, [1]/[t-BuOK] = 20) was added to a solution of the monomer 1 (2.02 g, 10.0 mmol) in dry THF (4.50 mL, [1] = 2.0 mol L−1) at 50 °C using a microsyringe. After 600 h, the reaction mixture was poured into a large amount of methanol, then the solution was neutralized with dry ice. After evaporating the solvent, the residue was purified by reprecipitation from methanol–water to yield the crude polymer. The precipitate was redissolved in CHCl3 (15 mL) and applied to the preparative SEC to remove the unreacted monomer 1. The concentrated polymer solution was dried in vacuo to give a white solid polymer 2 in a 68.4% yield (1.38 g); Mw,SEC (THF) = 1400; Mw,SEC/Mn,SEC (THF) = 1.28; Mw,SEC-MALLS (THF) = 7400; Mw,SEC-MALLS/Mn,SEC-MALLS (THF) = 1.37; dn·dc−1 (THF) = 0.080 mL g−1; [α]D = −23.1° (c 1.00, CHCl3, 24 °C). 1H NMR (CD2Cl2):δ 5.87 (br.s, H-1), 4.80–3.20 (m, including peaks at 4.59, 4.48, 4.26, 4.01, 3.73, 3.60, and 3.47 ppm), 3.06 (br.s), 2.93–2.70 (m, epoxy group), 1.44 (s, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)2) and 1.28 ppm (s, C(CH3)2). 13C NMR (CD2Cl2):δ 112.14 (C(CH3)2), 105.74 (C-1), 105.44 (C-1), 85.55, 84.20, 82.31, 80.46, 74.65, 73.45, 68.60, 48.49 (epoxy group), 46.98 (epoxy group), 26.96 (C(CH3)2), and 26.35 ppm (C(CH3)2). It is hard to estimate the degree of branching value because the NMR peaks due to each branch structure were broadened and overlapped.
C(CH3)2) and 1.28 ppm (s, C(CH3)2). 13C NMR (CD2Cl2):δ 112.14 (C(CH3)2), 105.74 (C-1), 105.44 (C-1), 85.55, 84.20, 82.31, 80.46, 74.65, 73.45, 68.60, 48.49 (epoxy group), 46.98 (epoxy group), 26.96 (C(CH3)2), and 26.35 ppm (C(CH3)2). It is hard to estimate the degree of branching value because the NMR peaks due to each branch structure were broadened and overlapped.
| Entry |
Cat. |
Temp/°C |
Polymerization time/h |
Addition time/h |
yield (%) |
M
w,SEC (Mw/Mn) c |
M
w,SEC-MALLS (Mw/Mn) d |
α
|
[η] d/mL g−1 |
K d/mL g−1 |
|
Conditions: Ar atmosphere; [M]/[I] = 20; solvent, THF; [M] = 2.0 mol L−1.
Condition: Ar atmosphere; [M]/[I] = 40; solvent, CH2Cl2; [M] = 0.5 mol L−1.
Determined by SEC using polystyrene standards in THF.
The weight average molecular weight Mw,SEC-MALLS, polydispersity Mw,SEC-MALLS/Mn,SEC-MALLS, intrinsic viscosity [η], and Mark–Houwink–Sakurada constants, α and K ([η] = KMw,SEC-MALLSα), were determined by SEC in THF equipped with MALLS and viscosity detectors.
|
| 1 |
t-BuOKa |
60 |
160 |
— |
52.9 |
2000 (1.26) |
7700 (1.22) |
0.27 |
4.6 |
0.40 |
| 2 |
t-BuOKa |
50 |
600 |
— |
68.4 |
1400 (1.28) |
7400 (1.37) |
0.25 |
4.0 |
0.48 |
| 3 |
t-BuOKa |
50 |
770 |
— |
69.5 |
1600 (1.34) |
8900 (1.33) |
0.27 |
4.3 |
0.38 |
| 4 |
BF3·OEt2b |
10 |
24 |
— |
68.5 |
4100 (1.29) |
15000 (1.45) |
0.11 |
3.4 |
1.16 |
| 5 |
BF3·OEt2b |
10 |
24 |
4.4 |
73.4 |
5600 (1.32) |
37000 (1.19) |
0.09 |
3.3 |
1.22 |
| 6 |
BF3·OEt2b |
10 |
24 |
13.3 |
75.5 |
6500 (1.33) |
81300 (1.23) |
0.09 |
3.4 |
1.26 |
| 7 |
BF3·OEt2b |
10 |
24 |
17.8 |
66.1 |
8600 (1.26) |
122400 (1.70) |
0.08 |
3.8 |
1.42 |
All procedures were performed using a Schlenk flask under an argon atmosphere. A typical procedure for the cationic polymerization (entry 4 in Table 1) is as follows: BF3·OEt2 (8.0 μL, 62.5 μmol, [1]/[BF3·OEt2] = 40) was added to a solution of 1 (0.505 g, 2.50 mmol) in dry CH2Cl2 (5.0 mL, [1] = 0.5 mol L−1) at 10 °C using a microsyringe. After 24 h, the polymerization was terminated by adding methanol containing a small portion of an aqueous ammonia solution. After evaporating the solvent, the residue was purified by reprecipitation from methanol–water to yield the crude polymer. The precipitate was redissolved in CHCl3 (4 mL) and applied to the preparative SEC to remove the unreacted monomer 1. The concentrated polymer solution was dried in vacuo to give a white solid polymer 2 in a 68.5% yield (346 mg); Mw,SEC (THF) = 4100; Mw,SEC/Mn,SEC (THF) = 1.29; Mw,SEC-MALLS (THF) = 15000; Mw,SEC-MALLS/Mn,SEC-MALLS (THF) = 1.45; dn·dc−1 (THF) = 0.079 mL g−1; [α]D = −50.6° (c 1.00, CHCl3, 24 °C). The 1H and 13C NMR spectra of 2 (entry 4) are almost similar to those of entry 7.
A typical procedure for the cationic polymerization (entry 7 in Table 1) using the slow-monomer-addition strategy87 is as follows: BF3·OEt2 (32.0 μL, 250 μmol, [1]/[BF3·OEt2] = 40) was added to a solution of 1 (0.404 g, 2.00 mmol) in dry CH2Cl2 (4.0 mL, [1] = 0.5 mol L−1) at 10 °C using a microsyringe. A dry CH2Cl2 (16.0 mL, [1] = 0.5 mol L−1) solution of 1 (1.62 g, 8.00 mmol) in a microsyringe was then slowly added to the mixture at 10 °C using a dosing pump. The drip rate was 15.0 μL min−1, i.e., the addition time of 1 was 17.8 h. After 24 h, the polymerization was terminated by adding 400 mL of methanol containing a small portion of an aqueous ammonia solution. After evaporating the solvent, the residue was purified by reprecipitation from methanol–water to yield the crude polymer. The precipitate was redissolved in CHCl3 (15 mL) and applied to the preparative SEC to remove the unreacted monomer 1. The concentrated polymer solution was dried in vacuo to give a white solid polymer 2 in a 66.1% yield (1.34 g); Mw,SEC (THF) = 8,600; Mw,SEC/Mn,SEC (THF) = 1.26; Mw,SEC-MALLS (THF) = 122,400; Mw,SEC-MALLS/Mn,SEC-MALLS (THF) = 1.70; dn·dc−1 (THF) = 0.074 mL g−1; [α]D = −22.2° (c 1.00, CHCl3, 25 °C). 1H NMR (CD2Cl2):δ 5.87 (br.s, H-1), 5.20–2.40 (br, including peaks at 4.47, 4.28, 4.18, 3.99, 2.91(epoxy group), 2.81 (epoxy group), 2.66 and 2.63 ppm), 1.44 (s, C(CH3)2), 1.28 ppm (s, C(CH3)2), and 0.89 ppm. 13C NMR (CD2Cl2):δ 111.95 (C(CH3)2), 106.83 (C-1), 105.40 (C-1), 101.49, 86.00–63.00 (m, including peaks at 85.62, 84.24, 82.16, 79.80, 75.35, 72.03, 69.39, 68.38), 27.05 (C(CH3)2), and 26.42 ppm (C(CH3)2). It is hard to estimate the degree of branching value because the NMR peaks due to each branch structure were broadened and overlapped.
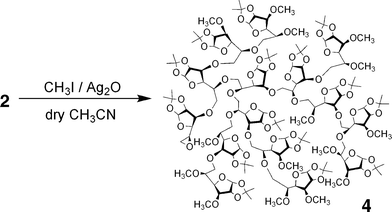
Silver oxide (0.50 g, 2.20 mmol) and methyl iodide (3.00 mL, 48.6 mmol) were added to polymer 2 (78.2 mg, 0.39 unit-mmol, entry 2 in Table 1) in 5 mL of dry acetonitrile. After the mixture was refluxed for 21 h, the insoluble parts were removed by filtration and the solvent was evaporated. The residue was redissolved in CHCl3 (1 mL) and applied to the preparative SEC to prepare the resulting polymer. The concentrated polymer solution was dried in vacuo to give a white solid polymer 4 in an 80.1% yield (67.0 mg); The degree of methylation, which was estimated from the 1H NMR measurement, was ca. 100%. Mw,SEC-MALLS (THF) = 4200; Mw,SEC-MALLS/Mn,SECMALLS (THF) = 1.49; dn·dc−1 (THF) = 0.076 mL g−1; [η] = 4.7 mL g−1; the Mark–Houwink–Sakurada exponents, α and K, are 0.22 and 0.90 mL g−1, respectively. 1H NMR (CD2Cl2):δ 5.83 (br.s, H-1), 4.55 (br.s), 4.30–3.20 (m, including peaks at 4.11, 3.93, 3.89, 3.77, 3.61, 3.60, 3.48 (OCH3), 3.46 (OCH3) and 3.44 ppm (OCH3)), 3.00–2.70 (m, including br.s at 2.91, 2.86, and 2.76 ppm, epoxy group) 1.45 (s, C(CH3)2), and 1.29 ppm (s, C(CH3)2). 13C NMR (CD2Cl2):δ 111.60 (C(CH3)2), 105.06 (C-1), 83.60–80.50 (m, including peaks at 83.51, 82.05, and 81.28 ppm), 78.50, 76.90–75.00 (m, including peak at 76.75 ppm), 74.00–67.00 (m, including peaks at 72.00, 69.97, and 67.50 ppm), 59.50–57.00 (m, including peaks at 58.43 and 57.64 ppm, OCH3), 48.15, 46.49, 29.68, 26.77 (C(CH3)2), and 26.28 ppm (C(CH3)2).
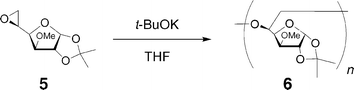
Synthesis of 5,6-anhydro-1,2-O-isopropylidene-3-O-methyl-α-D-glucofuranose (5)
Silver oxide (4.60 g, 19.8 mmol) and methyl iodide (1.20 mL, 19.8 mmol) were added to 1 (1.95 g, 9.64 mmol) in 5 mL of dry acetonitrile. After the mixture was refluxed for 20 h, the insoluble parts were removed by filtration and the solvent was evaporated. The residue was purified by column chromatography on silica gel with n-hexane/ethyl acetate (2/1, v/v; Rf 0.31) to afford colorless liquid 5. Yield: 1.92 g (92.1%). 1H NMR (CDCl3):δ 5.88 (d, J = 3.2 Hz, 1H, H-1), 4.58 (m, 1H, H-2), 3.82 (m, 1H, H-4), 3.68 (m, 1H, H-3), (s, 3H, OCH3), 3.46, 3.20 (m, 1H, H-5), 2.88 (m, 1H, H-6), 2.74 (m, 1H, H-6), 1.43 (s, 3H, C(CH3)2) and 1.29 ppm (s, 3H, C(CH3)2). 13C NMR (CDCl3):δ 111.9 (C(CH3)2), 105.3 (C-1), 84.4 (C-2), 82.0 (C-3), 81.6 (C-4), 58.4 (OCH3), 48.1 (C-5), 46.9 (C-6), 26.9 (C(CH3)2) and 26.3 ppm (C(CH3)2). Anal. Calcd for C10H16O5 (216.23): C, 55.55; H, 7.46. Found: C, 55.24; H, 7.31. FI-MS (m/z): 216 (M+).
The anionic polymerizations of 5 were carried out using a storage flask with a needle valve under an argon atmosphere. 1.0 mol L−1t-BuOK in a THF solution (0.30 mL, 0.30 mmol, [1]/[t-BuOK] = 20) was added to a solution of 5 (1.30 g, 6.00 mmol) in dry THF (2.70 mL, [1] = 2.0 mol L−1) at 60 °C using a microsyringe. After 180 h, the reaction mixture was poured into a large amount of methanol, and the solution was neutralized with dry ice. After evaporating the solvent, the residue was purified by reprecipitation from chloroform–n-hexane to yield the crude polymer. The precipitate was redissolved in CHCl3 (9 mL) and applied to the preparative SEC to remove the unreacted monomer. The concentrated polymer solution was dried in vacuo to give a white solid polymer, poly(5,6-anhydro-1,2-O-isopropylidene-3-O-methyl-α-D-glucofuranose) (6), in a 65.9% yield (857 mg); Mw,SEC-MALLS (THF) = 11100; Mw,SEC-MALLS/Mn,SEC-MALLS (THF) = 1.26; dn·dc−1 (THF) = 0.062 mL g−1; [η] = 6.8 mL g−1; Mark–Houwink–Sakurada exponents α and K are 0.50 and 0.06 mL g−1, respectively. [α]D = −51.0° (c 1.00, CHCl3, 25 °C). 1H NMR (CDCl3):δ 5.93 (d, J = 3.2 Hz, H-1), 5.83, 4.54, 4.49 (d, J = 3.7 Hz, H-2), 4.27 (d, J = 8.7 Hz, H-6), 4.01 (br.s, H-3 and H-4), 3.87, 3.76 (br.s, H-5), 3.53 (br.s, H-6) 3.46 (s, 3H, OCH3), 1.45 (s, C(CH3)2), 1.27 (s, C(CH3)2) and 1.17 ppm. 13C NMR (CDCl3):δ 111.03, 110.95 (C(CH3)2), 105.01 (C-1), 82.95 (C-4), 81.76 (C-2), 79.10 (C-3), 75.65 (C-5), 74.37 (C-6), 72.63, 57.61 (OCH3), 27.58, 26.76 (C(CH3)2) and 26.30 (C(CH3)2).
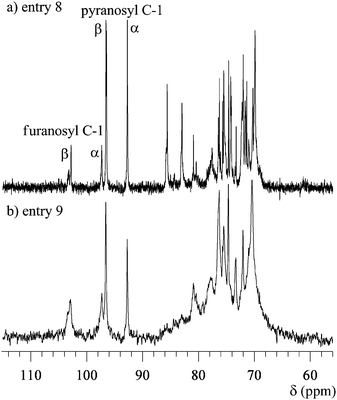
Polymer 2 (0.60 g, 2.97 unit-mmol, entry 7) was dissolved in 5 mL of a 85/15 (v/v) trifluoroacetic acid/water mixture. The solution was magnetically stirred at room temperature for 18 h, and diluted with 40 mL of water. The solution was neutralized using an aqueous ammonia solution, then dialyzed in water for several days using a Spectra/Por® CE (cellulose Ester) membrane (MWCO: 1000 Dalton, SPECTRUM). After freeze-drying from water, the residue was redissolved in water (5 mL) and applied to the preparative SEC to purify the resulting polymer. After freeze-drying from water, the hyperbranched 5,6-glucan (3) was isolated as a white powder. The yield was 62.7% (335 mg, entry 9). Mw,SEC-MALLS (0.2 mol L−1 NaNO3 aq.) = 74400; Mw,SEC-MALLS/Mn,SEC-MALLS (0.2 mol L−1 NaNO3 aq.) = 2.74; dn·dc−1 (0.2 mol L−1 NaNO3 aq.) = 0.135 mL g−1; [α]D = +17.3° (c 1.00, H2O, 25 °C). 1H NMR (entry 9, D2O including acetone as internal reference):δ 6.05 (br.s), 5.49 (br.s, α-franosyl unit, H-1), 5.23 (br.s, β-franosyl and α-pyranosyl units, H-1), and 5.00–3.20 ppm (m, including peaks at 4.64 (br.s, β-pyranosyl unit, H-1), 4.28, 4.12, 3.96, 3.71, 3.56, 3.48, 3.26), 1.44, and 1.23 ppm. 13C NMR (entry 9, D2O including acetone as an internal reference): δ 102.94 (β-franosyl unit, C-1), 97.30 (α-franosyl unit, C-1), 96.58 (β-pyranosyl unit, C-1), 92.74 (α-pyranosyl unit, C-1), and 88.00–62.00 ppm (m, including signals at 80.92, 77.83, 76.28, 75.48, 74.68, 73.33, 72.04, and 70.45 ppm). For entry 8: [α]D + 53.6° (c 1.00, H2O, 25 °C). 1H NMR (entry 8, D2O including acetone as internal reference):δ 6.01 (br.s), 5.48 (br.s, α-franosyl unit, H-1), 5.22 (br.s, β-franosyl and α-pyranosyl units, H-1), 4.65 (br.s, β-pyranosyl unit, H-1) and 4.50–3.20 ppm (m, including peaks at 4.33, 4.28, 4.12, 4.08, 3.99, 3.60, 3.52, 3.49, 3.47, 3.44, 3.40, 3.34, 3.33, 3.25), 1.52, and 1.36 ppm. 13C NMR (entry 8, D2O including acetone as internal reference): δ 103.24 (C-1), 102.80 (β-franosyl unit, C-1), 97.31 (α-franosyl unit, C-1), 96.56 (β-pyranosyl unit,C-1), 92.72 (α-pyranosyl unit, C-1), 85.62, 82.97, 80.89, 80.38, and 79.00–68.00 ppm (m, including signals at 77.65, 76.43, 76.25, 76.05, 75.68, 75.49, 74.62, 74.24, 74.15, 73.27, 72.21, 72.01, 71.68, 71.60, 71.37, 71.03, 70.56, and 69.90 ppm).
A typical procedure for the formation of the gold particles is as follows (run 1 in Fig. 15): polymer 3 (37.5 mg, 0.231 unit-mmol, entry 8) was first dissolved in deionized (Mill-Q) water (5 mL). The polymer solution (46 unit-mmol L−1, 1.8 mL) was filtered, then kept at 70 °C for 30 min in the UV-cell equipped with a stirring bar. The tetrachloroauric acid (HAuCl4) aqueous solution (0.01 mol L−1, 6.67 μL) was added to the polymer solution. The UV-vis spectra of the polymer–metal solution were measured in the wavelength range of 450–600 nm at 1 min intervals with the polymer solution as a reference. The obtained absorbance data were corrected by removing the absorption of the HAuCl4 aqueous solution.
Results and discussion
The ring-opening polymerizations of 5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose (1) were carried out using potassium tert-butoxide (t-BuOK) as the anionic initiator or boron trifluoride diethyletherate (BF3·OEt2) as the cationic initiator. The typical results are summarized in Table 1. When t-BuOK was used in dry tetrahydrofuran (THF) as the solvent, the reaction mixtures immediately changed from colorless to dark brown as the reaction progressed, and the polymerization homogeneously proceeded until the end of the polymerization, while the cationic polymerization using BF3·OEt2 in dry methylene chloride (CH2Cl2) homogeneously proceeded without any color change. The polymers obtained using t-BuOK or BF3·OEt2 were a light-yellow solid or a white solid, respectively, which were soluble in chloroform, THF, DMF and methanol, but insoluble in water. The polymer yields for the anionic polymerization (entries 1–3) were in the range of 52.9 to 69.5% and the increase in conversion was very slow, e.g., 52.9% at 60 °C for 160 h (entry 1), 68.4% at 50 °C for 600 h (entry 2) and 69.5% at 50 °C for 770 h (entry 3). The weight-average molecular weights (Mw,SEC) measured by size exclusion chromatography (SEC) of the resulting polymers ranged from approximately 1400 to 2000. In contrast, the polymer yield for the cationic polymerization (entry 4) was 68.5% for 24 h and the Mw,SEC was 4100, indicating that the cationic polymerization is much better than the anionic one.88
In general, highly branched polymers are known to have spherical conformations in a solution, and the Mw,SECs of these polymers were often claimed to be too low because the hydrodynamic volumes of these polymers are smaller than the corresponding linear polymers used for the calibration. Therefore, the absolute weight-average molecular weight (Mw,SEC-MALLS) of the resulting polymers was also measured using SEC equipped with a multiangle laser light scattering instrument (MALLS), and was compared with the Mw,SEC values in Table 1. The Mw,SEC-MALLS values for entries 1–4 ranged from 7400 to 15000, which were ca. 1.6–5.5 those of the Mw,SEC values. These results suggest that the resulting polymer has a compact form in solution because of its branched structure.
From the viscosity measurement in THF using SEC equipped with the viscosity detector, the intrinsic viscosities ([η]) of the resulting polymers were observed. The solution viscosities of the polymers obtained using t-BuOK were very low in the range from 4.0 to 4.6 mL g−1. The Mw,SEC-MALLS dependence of [η] for the polymer (entry 2) is shown in Fig. 2. On the basis of these results, the exponents (α) of the Mark–Houwink–Sakurada equations ([η] = KMα) of the polymers obtained using t-BuOK were determined to be 0.25–0.27. In general, α = 0.5 suggests that the polymer behaves as dense spheres, α = 0.6–0.8 for a flexible chain and α > 1 for an elongated rod, whereas it is well-known that the α value was less than 0.5 for the various hyperbranched polymers. Our experimental α values were noticeably low, which are ascribed to the compact spherical structure in solution. Therefore, the SEC, MALLS, and viscosity measurement results suggested that the polymer obtained using t-BuOK was a branched spherical molecule, i.e., a hyperbranched poly(5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose) (2).
![Double logarithmic plots of [η] vs.Mw,SEC-MALLS for polymer 2 in THF (entry 2).](/image/article/2010/PY/b9py00223e/b9py00223e-f2.gif) |
| | Fig. 2 Double logarithmic plots of [η] vs.Mw,SEC-MALLS for polymer 2 in THF (entry 2). | |
The solution viscosity of the polymer (entry 4) obtained using BF3·OEt2 was 3.4 mL g−1, which was lower than those of the polymers obtained using t-BuOK, regardless of the higher molecular weight polymer. In addition, the α value was 0.11, which was less than those of the polymers obtained using t-BuOK. These results indicate that the polymer obtained using BF3·OEt2 should have a more compact spherical structure in solution than the polymer obtained using t-BuOK, i.e., the polymer obtained using BF3·OEt2 should have a higher degree of branching value than the polymer obtained using t-BuOK.
As an efficient preparation method for hyperbranched polymers with both a high molecular weight and a high branching value degree, the slow-monomer-addition strategy87 was used for the cationic polymerization of 1. According to the established time of monomer addition, 16 mL of the 0.5 mol L−1 monomer solution was slowly added to the mixture of 4 mL of monomer solution and BF3·OEt2 at 10 °C using a dosing pump. These results are summarized in entries 5–7 of Table 1. The slow-monomer-addition strategy results in a very low monomer concentration actually present in the reaction mixture. Thus, the exclusive reaction of the monomer with the growing polyfunctional macromolecules occurs, resulting in a high molecular weight and a high degree of branching. Based on a longer addition time, i.e., a decrease in the drop rate, the Mw,SEC-MALLS values dramatically increase up to 122,400 (entry 7). Interestingly, regardless of the higher molecular weight polymer, the [η] values were in the range from 3.3 to 3.8 mL g−1, which was similar to the value for entry 4. The α values of the Mark–Houwink–Sakurada equations were determined to be very low in range of 0.08 to 0.09, suggesting that the resulting polymers have a highly branched spherical structure. In addition, the polydispersities (Mw,SEC/Mn,SEC) were found to be in the relatively narrow range of 1.26–1.33. These results indicate that the slow-monomer-addition strategy is a facile method for preparing the hyperbranched polymer 2 with a controlled molecular weight and highly branched structure.
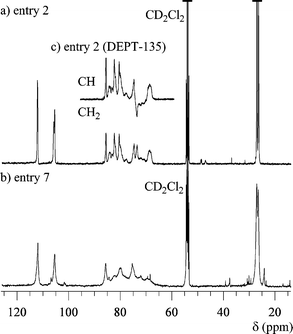
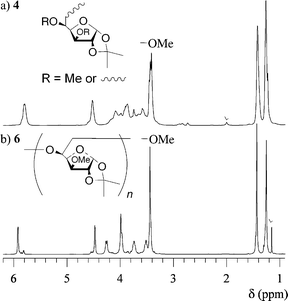
In order to investigate the polymer structure, the 1H and 13C NMR measurements were carried out in CD2Cl2. Parts a and b of Fig. 3 show the 1H NMR spectra of the polymers obtained using t-BuOK (entry 2) and BF3·OEt2 (entry 7), respectively. Though the unreacted monomer was removed using the preparative SEC, in both spectra, the peaks due to the epoxy groups slightly exist in the range of 2.6–3.0 ppm as the starting polymer end, which is a characteristic of a hyperbranched polymer. From a comparison of both spectra, the spectrum of the polymer obtained using t-BuOK is obviously different from that of the polymer using BF3·OEt2, recognizing that the structure of the former is more regular than that of the latter because of distinct splitting of the peaks and narrow peak width for the anionically obtained polymer. This trend is also recognized by the comparison of the 13C NMR spectra in Fig. 4. Ring-opening of the monosubstituted epoxide occurs in two ways, i.e., by α- or β-scission. In general, the polymerization using an anionic catalyst predominantly cleaves the CH2–O bond (β-scission) via an SN2 displacement to form the regular head-to-tail linkage. Therefore, the difference in the spectra is mainly caused by the stereoregularity of the C-5 carbon in the polymer. Though the anionically obtained polymer has higher stereoregularity, the 13C NMR spectrum contains many peaks due to the C-6 methylene carbons, as shown in Fig. 4c, indicating that the polymer obviously contains some branched structures as irregular structure.
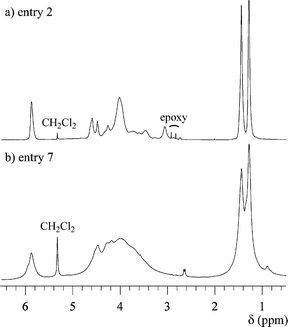 |
| | Fig. 3
1H NMR spectra of polymer 2 in CD2Cl2 (TMS as the internal standard), (a) entry 2 and (b) entry 7. | |
In order to investigate the branching structure in polymer 2, the 1H and 13C NMR spectra of the methylated hyperbranched polymer (4) were compared with those of the linear analog, poly(5,6-anhydro-1,2-O-isopropylidene-3-O-methyl-α-D-glucofuranose) (6). The polymer 4 was prepared by the methylation of the anionically obtained polymer 2 (entry 2) using CH3I and Ag2O in dry CH3CN, as shown in Fig. 5. On the other hand, the linear polymer 6 was obtained by the anionic polymerization of 5,6-anhydro-1,2-O-isopropylidene-3-O-methyl-α-D-glucofuranose (5) using t-BuOK, as shown in Fig. 6. Parts a and b of Fig. 7 show the 1H NMR spectra of 4 and 6, respectively. The 1H NMR spectrum of 4 consists of some broad peaks, which is obviously different from that of 6. In particular, the methoxy peaks of 4 were split into three or more, indicating that there are some methoxy groups in different surroundings, as shown in Fig. S1 (see ESI†). A comparison of the 13C NMR spectra in Fig. S2 (see ESI†) also shows the obvious difference. The 13C NMR spectrum of 6 mainly consists of 10 sharp peaks corresponding to the repeating unit, while the spectrum of 4 includes some broad peaks which are due to the irregular structure. In addition, the Mark–Houwink–Sakurada exponent α of 4 was 0.22, whereas the α value of 6 was 0.50. The NMR and viscosity results strongly support the fact that polymer 4 obviously had some branched structure as an irregular structure, i.e., the polymerization of 1 proceeded through the ring-opening multibranching mechanism to produce the hyperbranched poly(5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose) 2.
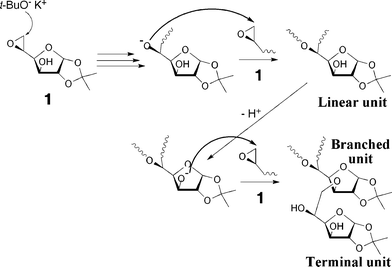
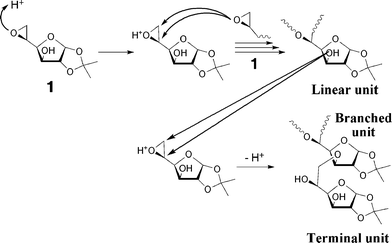
Fig. 8 and 9 represent the proposed anionic and cationic polymerization mechanisms, respectively. During the anionic polymerization, the ring-opening occurred through only β-scission to afford the linear unit with the regular head-to-tail linkage. The branches of the polymers should be produced by the proton transfer from the hydroxy group at C-3 to the other alkoxide and the following ring opening of 1 by the alkoxide at C-3. On the other hand, for the cationic polymerization, the protonated 1 reacted with the epoxy group of a second monomer, and two kinds of linkages through the α- or β-scission were formed as a linear structure. The branched structure should be produced by proton transfer from the hydroxyl groups in the polymer chains to the oxygen atom of the epoxy group. These reactions mentioned above simultaneously occurred in the polymerization systems and consequently produced the hyperbranched polymer 2.
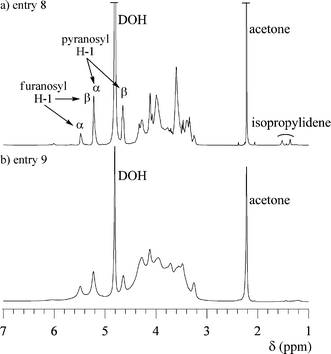
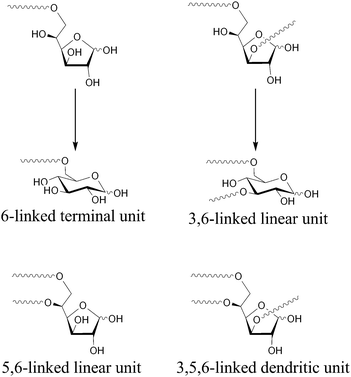
The hydrolysis of polymer 2 was carried out using a mixture of trifluoroacetic acid and water (85/15, v/v) to produce the hyperbranched poly(5,6-anhydro-α-D-glucofuranose) (3), e.g., the hyperbranched 5,6-glucan. The obtained polymers 3 were white powders which were soluble in water, and insoluble in chloroform, THF, DMF and methanol, which remarkably differed from the solubility of 2. Fig. 10 and 11 show the 1H and 13C NMR spectra of 3 (entries 8 and 9) obtained from 2 (entries 2 and 7), respectively. Based on these spectra, it is concluded that the isopropylidene groups were mostly hydrolyzed. The physical data of the obtained 3 are listed in Table 2. The Mw,SEC-MALLS values for entries 8 and 9 are 3740 and 74400, respectively, which were lower than the calculated Mw values. This indicates that some linkage may break under the hydrolysis conditions. However, the exponents, α, of the Mark–Houwink–Sakurada equations were determined to be 0.36 for entry 8 and 0.26 for entry 9, as shown in Fig. 12, indicating that the obtained polymer still have the branched structure. The 1H and 13C NMR spectra of 3 consisted of very complicated peaks, which were constructed by the difference sequences of the D-glucose units. In the 13C NMR spectra, four distinct signals exist in the C-1 region. The big signals at 92.74 and 96.58 ppm in Fig. 11a, which are very similar to the chemical shifts of the carbons for α- and β-D-glucopyranoses,89 should be assigned to the carbons of the α- and β-D-glucopyranose units in 3, respectively. In addition, the small signals at 97.30 and 102.94 ppm should be assigned to the carbons of the α- and β-D-glucofranose units in 3, respectively, since these signals are very similar to the chemical shifts of 3,5,6-tri-O-methyl α- and β-D-glucofranoses.90 This result indicates that after hydrolysis, most of the D-glucofranose units in 2 changes to the D-glucopyranose unit in 3. As shown in Fig. 13, the D-glucofranose units with a hydroxyl group at C-5 could isomerize to the D-glucopyranose units, e.g., the 6-linked terminal and 3,6-linked linear D-glucofranose units isomerize to the corresponding D-glucopyranose units, while the 5,6-linked linear and 3,5,6-linked dendritic units do not change. Since the intensities of the D-glucopyranose units in Fig. 11 are stronger than those of D-glucofranose, polymer 3 includes numerous 6-linked terminal and 3,6-linked linear D-glucopyranose units, which is a characteristic of a hyperbranched polymer.
Table 2 The results of hydrolysis of 2 to prepare 3
| Entry |
Starting material 2 |
M
w,SEC-MALLS of 3a (Mw,SEC-MALLS/Mn,SEC-MALLS) |
α
|
[η] a/mL g−1 |
K
/mL g−1 |
|
The weight average molecular weight Mw,SEC-MALLS, polydispersity Mw,SEC-MALLS/Mn,SEC-MALLS, intrinsic viscosity [η], and Mark–Houwink–Sakurada constants, α and K ([η] = KMw,SEC-MALLSα), were determined by SEC in 0.2 mol L−1 NaNO3 aqueous solution equipped with MALLS and viscosity detectors.
|
| 8 |
entry 2 |
3740 (1.26) |
0.36 |
4.4 |
0.22 |
| 9 |
entry 7 |
74400 (2.74) |
0.26 |
5.0 |
0.29 |
![Double logarithmic plots of [η] vs.Mw,SEC-MALLS for polymer 3 in 0.2 mol L−1 NaNO3 aqueous solution, (a) entry 8 and (b) entry 9.](/image/article/2010/PY/b9py00223e/b9py00223e-f12.gif) |
| | Fig. 12 Double logarithmic plots of [η] vs.Mw,SEC-MALLS for polymer 3 in 0.2 mol L−1 NaNO3 aqueous solution, (a) entry 8 and (b) entry 9. | |
To estimate the reducing ability of 3, the formation of gold particles, which is derived from the reduction of HAuCl4, is demonstrated in the aqueous solution of 3. Fig. 14 shows the UV-vis spectra after 150 min mixing of 3 and HAuCl4 aqueous solutions at 70 °C (runs 1–3). As a comparative experiment, the UV-vis spectrum of the mixture of D-glucose and HAuCl4 aqueous solutions (run 4) is also shown in Fig. 14.91 After mixing of the 3 and HAuCl4 aqueous solutions, the color of the system changed from colorless to light red with time, as shown in Fig. S3 (see ESI†). The UV-vis spectra of the 3-HAuCl4 system have a λmax that ranged from 512 nm to 514 nm, which due to the formation of the gold particles. From previous studies,92 the average gold particle diameter can be estimated from the λmax, i.e., gold particles of ca. 8–12 nm were obtained by the mixing of 3 and the HAuCl4 aqueous solutions. Thus, the average hydrodynamic diameter of the resulting Au particles (run 2 in Fig. 14), which was measured by a dynamic light scattering (DLS) technique, was ca. 11.3 nm, as shown in Fig. S4 (see ESI†).
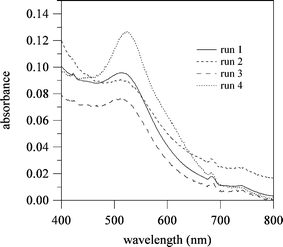 |
| | Fig. 14 UV-vis spectra after 150 min mixing of polymer 3 (or D-glucose) and HAuCl4 at 70 °C. Run 1: polymer 3 (46 unit-mmol L−1 of entry 8, 1.8 mL) and HAuCl4 (0.01 mol L−1, 6.67 μL) aqueous solutions; run 2: polymer 3 (25 unit-mmol L−1 of entry 8, 1.8 mL) and HAuCl4 (0.01 mol L−1, 6.67 μL) aqueous solutions; run 3: polymer 3 (46 unit-mmol L−1 of entry 9, 1.8 mL) and HAuCl4 (0.01 mol L−1, 6.67 μL) aqueous solutions; and run 4: D-glucose (46 unit-mmol L−1, 1.8 mL) and HAuCl4 (0.01 mol L−1, 6.67 μL) aqueous solutions. The maximal wavelengths (λmax) are 512 nm for run 1, 514 nm for run 2, 514 nm for run 3 and 526 nm for run 4. | |
To obtain the relationship between the formation of the gold particles and the reaction time, the increasing trend in the UV-vis absorbance at λmax is presented in Fig. 15. For all the 3-HAuCl4 systems (runs 1–3), the UV-vis absorbance sharply increases without an induction time and finally became a constant value within 10 min.93,94 On the other hand, the D-glucose-HAuCl4 system (run 4) shows a long induction time, then the absorbance sharply increases as an autocatalytic system. A similar autocatalytic tendency with a long induction time was also reported using the sugar-persubstituted poly(amidoamine) dendrimer, i.e., a sugar ball with numerous nonreducing sugar units.95 The results indicate that 3 has a high reducing ability that originated from the high density of the reducing sugars in 3, i.e., a glyco-cluster effect or multivalent effect of the reducing sugars. Thus, the reaction rates within 10 min for the 3-HAuCl4 systems were much faster than that for the D-glucose-HAuCl4 system, as shown in Fig. 15b. This indicates that “reducing sugar ball” 3 effectively act as a sugar based reductant.
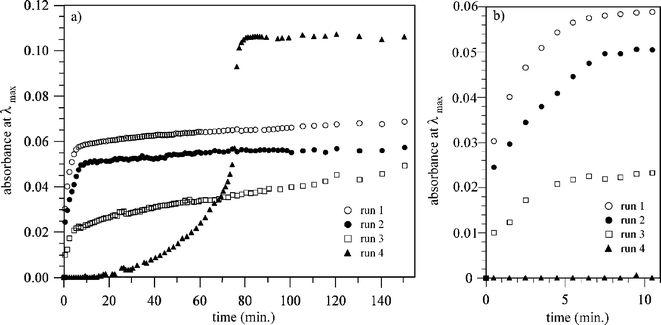 |
| | Fig. 15 The change in the UV absorption at λmax for mixtures of polymer 3 (or D-glucose) and HAuCl4 as a function of reaction time at 70 °C. Run 1 (○): polymer 3 (46 unit-mmol L−1 of entry 8, 1.8 mL) and HAuCl4 (0.01 mol L−1, 6.67 μL) aqueous solutions at 512 nm, run 2 (●): polymer 3 (25 unit-mmol L−1 of entry 8, 1.8 mL) and HAuCl4 (0.01 mol L−1, 6.67 μL) aqueous solutions 514 nm, run 3 (□): polymer 3 (46 unit-mmol L−1 of entry 9, 1.8 mL) and HAuCl4 (0.01 mol L−1, 6.67 μL) aqueous solutions 514 nm, and run 4 (▲): D-glucose (46 unit-mmol L−1, 1.8 mL) and HAuCl4 (0.01 mol L−1, 6.67 μL) aqueous solutions 526 nm. (a) Overall figure and (b) the expanded figure in the range of 0 to 10 min. The obtained absorbance data were corrected by removing the absorption of the HAuCl4 aqueous solution. | |
Conclusions
In this study, we performed the anionic or cationic ring-opening multibranching polymerization of 5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose (1) as a latent cyclic AB2-type monomer. The polymerizations proceeded through the proton-transfer reaction mechanism to produce hyperbranched poly(5,6-anhydro-1,2-O-isopropylidene-α-D-glucofuranose) (2). In particular, the cationic polymerization with the slow-monomer-addition strategy is a facile method leading to the polymers with a high molecular weight and highly-branched structure. The weight-average molecular weight (Mw,SEC-MALLS) values of 2 measured by multi-angle laser light scattering (MALLS) varied in the range of 7400 to 122400, which were significantly higher than the weight-average molecular weight (Mw,SEC) values by size exclusion chromatography (SEC). The intrinsic viscosities ([η]) of these polymers were very low in the range of 3.3–4.6 mL g−1 and the Mark–Houwink–Sakurada exponent α was calculated to be 0.08–0.28. These results of the MALLS, SEC, and viscosity measurements suggested that these polymers exist in a compact spherical conformation in solution because of their highly-branched structure. A water-soluble hyperbranched 5,6-glucan (3) was also synthesized by the hydrolysis of polymer 2. Polymer 3 is a novel hyperbranched glycopolymer arranged with numerous reducing D-glucose units on the peripheries of the polymer, and has a high reducing ability derived from the glycol-cluster effect or the multivalent effect of the reducing D-glucose units, i.e., the “reducing sugar ball” 3 effectively acts as a sugar-based reductant. Such a hyperbranched glycopolymer with numerous reducing sugar units can be expected to be useful in a wide variety of fields such as biochemical and medicinal applications; for example, a biocompatible reductant, antioxidant, and a part of the carrier for a drug delivery system, etc.
Acknowledgements
This study was supported by the Industrial Technology Research Grant Program in 2005 from the New Energy and Industrial Technology Development Organization (NEDO) of Japan and the Research Fellowships of the Japan Society for the Promotion of Science (JSPS) for Young Scientists. The authors thank the OPEN FACILITY (Hokkaido University Sousei Hall) for the MALLS (THF) and DLS measurements.
References
- C. J. Hawker and W. Devonport, ACS Symp. Ser., 1996, 624, 186 CAS.
- J. M. J. Fréchet, C. J. Hawker, I. Gitsov and J. W. Leon, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 33, 1399 Search PubMed.
- F. Zeng and S. C. Zimmerman, Chem. Rev., 1997, 97, 1681 CrossRef CAS.
- O. A. Matthews, A. N. Shipway and J. F. Stoddart, Prog. Polym. Sci., 1998, 23, 1 CrossRef.
- Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS.
- A. Hult, M. Johansson and E. Malmström, Adv. Polym. Sci., 1999, 143, 1 CAS.
- M. Fischer and F. Vogtle, Angew. Chem., Int. Ed., 1999, 38, 884 CrossRef.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505 CrossRef CAS.
- F. Vogtle, S. Gestermann, R. Hesse, H. Schwierz and B. Windisch, Prog. Polym. Sci., 2000, 25, 987 CrossRef CAS.
- K. Inoue, Prog. Polym. Sci., 2000, 25, 453 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747 CrossRef CAS.
- M. Jikei and M.-A. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233 CrossRef CAS.
- H. Frey and R. Haag, Rev. Mol. Biotechnol., 2002, 90, 257 CrossRef CAS.
- S.-E. Stiriba, H. Frey and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 1329 CrossRef CAS.
- F. O. H. Pirrung, E. M. Loen and A. Noordam, Macromol. Symp., 2002, 187, 683 CrossRef CAS.
- W. B. Turnbull and J. F. Stoddart, Rev. Mol. Biotechnol., 2002, 90, 231 CrossRef CAS.
- H. Frey and R. Haag, Rev. Mol. Biotechnol., 2002, 90, 257 CrossRef CAS.
- D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719 CrossRef CAS.
-
I. Gitsov and K. R. Lambrych, Dendrimers. Synthesis and Applications, in Microspheres, Microcapsules & Liposomes, ed. R. Arshady, Citus Books, London, 2002, vol. 5, pp. 31–68 Search PubMed.
- S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1047 CrossRef CAS.
- K. Ishizu, K. Tsubaki, A. Mori and S. Uchida, Prog. Polym. Sci., 2003, 28, 27 CrossRef CAS.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS.
- A. Hirao, M. Hayashi, S. Loykulnant, K. Sugiyama, S. W. Ryu, N. Haraguchi, A. Matsuo and T. Higashihara, Prog. Polym. Sci., 2005, 30, 111 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487 CrossRef CAS.
- X. Feng, D. Taton, R. Borsali, E. L. Chaikof and Y. Gnanou, J. Am. Chem. Soc., 2006, 128, 11551 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS.
- T. Satoh, M. Tamaki, Y. Kitajyo, T. Maeda, H. Ishihara, T. Imai, H. Kaga and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 406 CrossRef CAS.
- A. Hirao, K. Sugiyama and H. Yokoyama, Prog. Polym. Sci., 2007, 32, 1393 CrossRef CAS.
- Y. Kitajyo, T. Imai, Y. Sakai, M. Tamaki, H. Tani, K. Takahashi, A. Narumi, H. Kaga, N. Kaneko, T. Satoh and T. Kakuchi, Polymer, 2007, 48, 1237 CrossRef CAS.
- Y. Kitajyo, Y. Nawa, M. Tamaki, H. Tani, K. Takahashi, H. Kaga, T. Satoh and T. Kakuchi, Polymer, 2007, 48, 4683 CrossRef CAS.
- S. Peleshanko and V. V. Tsukruk, Prog. Polym. Sci., 2008, 33, 523 CrossRef CAS.
- T. Satoh, Soft Matter, 2009, 5, 1972 RSC.
- K. L. Wooley, J. M. J. Fréchet and C. J. Hawker, Polymer, 1994, 35, 4489 CrossRef CAS.
- C. Schipp, M. J. Hill, P. J. Barham, V. M. Cloke, J. S. Higgins and L. Oiarzabalt, Polymer, 1996, 37, 2291 CrossRef CAS.
- C. J. Hawker, E. Malmström, C. W. Frank and J. P. Kampf, J. Am. Chem. Soc., 1997, 119, 9903 CrossRef CAS.
- M. W. Weimer, J. M. J. Fréchet and I. Gitsov, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 955 CrossRef CAS.
- P. G. Santangelo and C. M. Roland, Macromolecules, 1999, 32, 1972 CrossRef CAS.
- J.-I. Kadokawa, H. Tagaya and K. Chiba, Polym. Drugs Drug Delivery Syst., 2001, 251 Search PubMed.
- S.-E. Stiriba, H. Kautz and H. Frey, J. Am. Chem. Soc., 2002, 124, 9698 CrossRef CAS.
- M. Ballauff and C. N. Likos, Angew. Chem., Int. Ed., 2004, 43, 2998 CrossRef CAS.
- K. R. Kumar and D. E. Brooks, Macromol. Rapid Commun., 2005, 26, 155 CrossRef CAS.
- F. Samadi, J. Eckelt, B. A. Wolf, F.-J. Lopez-Villanueva and H. Frey, Eur. Polym. J., 2007, 43, 4236 CrossRef CAS.
- Y. Kitajyo, Y. Kinugawa, M. Tamaki, H. Kaga, N. Kaneko, T. Satoh and T. Kakuchi, Macromolecules, 2007, 40, 9313 CrossRef CAS.
- Y. Haba, C. Kojima, A. Harada and K. Kono, Angew. Chem., Int. Ed., 2007, 46, 234 CrossRef CAS.
- Y. Chi, S. T. Scroggins and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 6322 CrossRef CAS.
- S. Javor, A. Natalello, S. M. Doglia and J.-L. Reymond, J. Am. Chem. Soc., 2008, 130, 17248 CrossRef CAS.
- K. Aoi, K. Itoh and M. Okada, Macromolecules, 1995, 28, 5391 CrossRef CAS.
- K. Aoi, K. Tsutsumiuchi, A. Yamamoto and M. Okada, Tetrahedron, 1997, 53, 15415 CrossRef CAS.
- K. Aoi, K. Tsutsumiuchi, A. Yamamoto and M. Okada, Macromol. Rapid Commun., 1998, 19, 5 CrossRef CAS.
- D. Zanini and R. Roy, J. Org. Chem., 1998, 63, 3486 CrossRef CAS.
- P. R. Ashton, E. F. Hounsell, N. Jayaraman, T. M. Nilsen, N. Spencer, J. F. Stoddart and M. Young, J. Org. Chem., 1998, 63, 3429 CrossRef CAS.
- H. W. I. Peerlings, S. A. Nepogodiev, J. F. Stoddart and E. W. Maijer, Eur. J. Org. Chem., 1998, 1879 CAS.
- K. Tsutsumiuchi, K. Aoi and M. Okada, Polym. J., 1999, 31, 935 CrossRef CAS.
- S. André, P. J. C. Ortega, M. A. Perez, R. Roy and H.-J. Gabius, Glycobiology, 1999, 9, 1253 CrossRef CAS.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555 CrossRef CAS.
- S. M. Rele, W. Cui, L. Wang, S. Hou, G. Barr-Zarse, D. Tatton, Y. Gnanou, J. D. Esko and E. L. Chaikof, J. Am. Chem. Soc., 2005, 127, 10132 CrossRef CAS.
- H. Baigude, K. Katsuraya, S. Tokunaga, N. Fujiwara, M. Satoyama, T. Magome, K. Okuyama, G. Borjihan and T. Uryu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2195 CrossRef CAS.
- A. Takasu, T. Makino and T. Hirabayashi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 310 CrossRef CAS.
- B. W. Greatrex, S. J. Brodie, R. H. Furneaux, S. M. Hook, W. T. McBurney, G. F. Painter, T. Rades and P. M. Rendle, Tetrahedron, 2009, 65, 2939 CrossRef CAS.
- N. Jayaraman, S. A. Nepogodiev and J. F. Stoddart, Chem.–Eur. J., 1997, 3, 1193 CrossRef CAS.
- B. Colonna, V. D. Harding, S. A. Nepogodiev, F. M. Raymo, N. Spencer and J. F. Stoddart, Chem.–Eur. J., 1998, 4, 1244 CrossRef CAS.
- W. B. Turnbull, A. R. Pease and J. F. Stoddart, ChemBioChem, 2000, 1, 70 CrossRef CAS.
- W. B. Turnbull, S. A. Kalovidouris and J. F. Stoddart, Chem.–Eur. J., 2002, 8, 2988 CrossRef CAS.
- C. D. Heidecke and T. K. Lindhorst, Chem.–Eur. J., 2007, 13, 9056 CrossRef CAS.
- J. Bernard, A. Favier, L. Zhang, A. Nilasaroya, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2005, 38, 5475 CrossRef CAS.
- J. Bernard, X. Hao, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Biomacromolecules, 2006, 7, 232 CrossRef CAS.
- A. Narumi, S. Yamane, Y. Miura, H. Kaga, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4373 CrossRef CAS.
- A. Narumi, H. Kaga, Y. Miura, T. Satoh, N. Kaneko and T. Kakuchi, Polymer, 2006, 47, 2269 CrossRef CAS.
- A. Narumi, H. Kaga, Y. Miura, I. Otsuka, T. Satoh, N. Kaneko and T. Kakuchi, Biomacromolecules, 2006, 7, 1496 CrossRef CAS.
- A. Narumi and T. Kakuchi, Polym. J., 2008, 40, 383 CrossRef CAS.
- J.-I. Kadokawa, M. Sato, M. Karasu, H. Tagaya and K. Chiba, Angew. Chem., Int. Ed., 1998, 37, 2373 CrossRef CAS.
- A. Kanazawa, S. Okumura and M. Suzuki, Org. Biomol. Chem., 2005, 3, 1746 RSC.
- S. Muthukrishnan, G. Jutz, X. Andre, H. Mori and A. H. E. Müller, Macromolecules, 2005, 38, 9 CrossRef CAS.
- S. Muthukrishnan, H. Mori and A. H. E. Müller, Macromolecules, 2005, 38, 3108 CrossRef CAS.
- S. Muthukrishnan, F. Plamper, H. Mori and A. H. E. Müller, Macromolecules, 2005, 38, 10631 CrossRef CAS.
- A. Kanazawa and M. Suzuki, Polymer, 2006, 47, 176 CrossRef CAS.
- T. Satoh, T. Imai, H. Ishihara, T. Maeda, Y. Kitajyo, A. Narumi, H. Kaga, N. Kaneko and T. Kakuchi, Macromolecules, 2003, 36, 6364 CrossRef CAS.
- T. Imai, T. Satoh, H. Kaga, N. Kaneko and T. Kakuchi, Macromolecules, 2003, 36, 6359 CrossRef CAS.
- T. Imai, T. Satoh, H. Kaga, N. Kaneko and T. Kakuchi, Macromolecules, 2004, 37, 3113 CrossRef CAS.
- T. Satoh, T. Imai, Y. Kitajyo, T. Maeda, A. Narumi, H. Kaga, N. Kaneko and T. Kakuchi, Macromol. Symp., 2004, 217, 39 CrossRef CAS.
- T. Imai, Y. Nawa, Y. Kitajyo, T. Satoh, H. Kaga, N. Kaneko and T. Kakuchi, Macromolecules, 2005, 38, 1648 CrossRef CAS.
- T. Satoh, T. Imai, H. Ishihara, T. Maeda, Y. Kitajyo, Y. Sakai, H. Kaga, N. Kaneko, F. Ishii and T. Kakuchi, Macromolecules, 2005, 38, 4202 CrossRef CAS.
- T. Satoh and T. Kakuchi, Macromol. Biosci., 2007, 7, 999 CrossRef CAS.
- T. Uryu, K. Kitano and K. Matsuzaki, Makromol. Chem., 1979, 180, 1135 CrossRef CAS.
- T. Uryu, K. Kitano, H. Tachikawa, K. Ito and K. Matsuzaki, Makromol. Chem., 1978, 179, 1773 CrossRef CAS.
- T. Soler, A. Bachki, L. R. Falvello, F. Foubelo and M. Yus, Tetrahedron: Asymmetry, 2000, 11, 493 CrossRef CAS.
- A. Sunder, R. Hanselmann and H. Frey, Macromolecules, 1999, 32, 4240 CrossRef CAS.
- A similar polymerization tendency was reported by Uryu et al.84,85.
- The chemical shifts of the carbons for α- and β-D-glucopyranoses in D2O are 93.18 and 97.00 ppm, respectively.
- G. Just and D. Crosilla, Can. J. Chem., 1980, 58, 2349 CAS The chemical shifts of the protons for 3,5,6-tri- O-methyl α- and β-D-glucofranoses in CDCl3 are 5.40 and 5.10 ppm, respectively. Based on the 1H NMR data of 3,5,6-tri- O-methyl α- and β-D-glucofranoses and CH-COSY spectrum of polymer 3 (entry 8) in Fig. S5 (see ESI†), the signals at 97.30 and 102.94 ppm in Fig. 11a are assigned to the carbons of the α- and β-D-glucofranose units in 3, respectively.
- J. Liu, G. Qin, P. Raveendran and Y. Ikushima, Chem.–Eur. J., 2006, 12, 2131 CrossRef CAS.
- K. Nakamura, T. Kawabata and Y. Mori, Powder Technol., 2003, 131, 120 CrossRef CAS.
- In run 3 using the high molecular weight polymer 3 (entry 9), the intensity at λmax is consistently rather weaker compared to run 1 using the low molecular weight sample (entry 8), probably because the sugar units on the peripheries of polymer 3 at the same unit-mol concentration is less for the high molecular weight polymer than the low molecular weight polymer, i.e., the inner sugar units of the high molecular weight polymer cannot effectively be utilized as a reductant because of the sugar units in the enclosed space.
- The final intensity at λmax for the D-glucose-HAuCl4 system was greater than those for the 3-HAuCl4 system at the same mol concentration. The main factor is attributed to the links between the sugar units in 3, which cannot act as a reductant.
- K. Esumi, T. Hosoya, A. Suzuki and K. Torigoe, J. Colloid Interface Sci., 2000, 226, 346 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.