Sequence-controlled polymerizations: the next Holy Grail in polymer science?
Jean-François
Lutz
*
Research Group Nanotechnology for Life Science, Fraunhofer Institute for Applied Polymer Research, Geiselbergstrasse 69, 14476, Potsdam-Golm, Germany. E-mail: lutz@iap.fhg.de; Fax: +49 331 568 3000; Tel: +49 331 568 1127
First published on 4th January 2010
Abstract
The aim of this short perspective article is to sensitize polymer chemists to the importance of controlling comonomer sequences. During the last twenty years, our scientific community has made impressive progress in controlling the architecture of synthetic macromolecules (i.e. chain length, shape and composition). In comparison, our tools for controlling polymer microstructures (i.e. sequences and tacticity) are still very rudimentary. However, as learned from Nature, sequence-controlled polymers are most likely the key toward functional sub-nanometric materials.
1. Introduction
Polymer-based materials have evolved drastically over the last years. In particular, the rapid emergence of polymer nanotechnology has opened up new possibilities for designing functional materials with tailored morphologies.1–4 For example, the self-organization of block copolymers in solution or in bulk leads to the formation of various types of nanostructured objects.5–11 Yet, in these nanoscale materials, the essential building-blocks are in most cases long homopolymer chains.12 This implies a precision in materials design of approximately 100–500 atoms. In contrast, Nature is able to control materials properties at a much lower scale. For instance, many proteins are constituted of a single polypeptide chain, which folds into a precise functional tertiary structure. Such folded macromolecular chains are able to perform advanced tasks (e.g. organocatalysis, selective transport, signal transduction), which are currently out of reach with standard synthetic polymers. To attain such a molecular precision, Nature hardly relies on polymers of controlled architecture (e.g. block copolymers) but rather on polymers with controlled microstructures (i.e. controlled sequences and tacticity).Hence, following Nature's example, it seems very plausible to achieve new generations of highly-organized materials using sequence-controlled polymers.13,14 For instance, several pioneering groups reported recently that natural biopolymers such as nucleic acids or peptides can be used to organize synthetic materials.15–20 These “bio-inspired” strategies opened interesting avenues for materials design. Yet, it is hard to imagine that the future of materials science will solely rely on biological polymers. In fact, it is possible to foresee with reasonable certainty that synthetic polymers with controlled monomer sequences can also play a significant role in that regard. However, in order to reach that goal, reliable methods for controlling monomer sequences have to be developed in the near future. This certainly implies an important collective research effort at the interface of polymer chemistry, polymer physico-chemistry and polymer physics.
In this scientific context, the goal of the present article is to re-stimulate the research on polymer sequences. In particular, this text is specifically written for polymer chemists. Thus, biological polymerizations (e.g. replication, transcription and translation) and the solid-phase synthesis of sequence-defined biopolymers (e.g. oligopeptide or oligonucleotide synthesis) are not covered in the present manuscript. However, some background information about these important processes can be found in a recent review.14 Herein, current status, future challenges, and promising trends in the field of polymer sequences are analyzed and discussed.
2. The current situation
The control over the primary structures (i.e. comonomer sequences) of synthetic macromolecules is certainly not a main topic in polymer science nowadays. Only a few articles on the subject have been published in leading polymer journals during the last decade. But this situation is relatively recent. Indeed, very important efforts have been made during the last century for understanding and characterizing monomer sequence distribution in chain-growth or step-growth polymerization processes. The state-of-the-art is briefly described in the following sub-sections (2.1–2.3).2.1 Step-growth polymerizations
Step-growth polymerizations rely on functional monomers containing reactive termini (x, y and z in Scheme 1). In solution (i.e. in a batch mode where a large amount of growing chains are coexisting) and in the absence of protecting chemistries, these polymerizations offer limited options for controlling primary monomer sequences. For example, the step-growth polymerization of distinct comonomers of the xy-type (i.e. each comonomer contains terminal x and y reactive functions) typically lead to statistical polymerizations (Scheme 1, top left). On the other hand, copolymerizations based on two comonomers xx and yy lead to ordered AB alternating sequences. This constitutes an interesting example of controlled primary structures obtained via a synthetic polymerization process.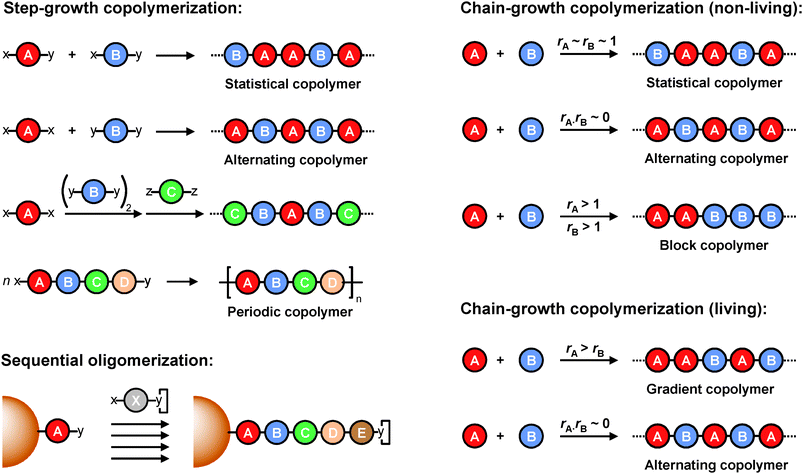 | ||
| Scheme 1 State of the art: microstructures attainable in standard synthetic polymerization processes. The capital letters A, B, C, D, and E represent different monomer units. The capital letter X represents any kind of monomer unit. The letters x, y, and z represent different types of reactive functions (x reacts with y; y reacts with z). The letter y included in a square box (bottom left) represents a protected y function. The symbols rA and rB represent the reactivity ratios of monomers A and B, respectively. | ||
Yet, this interesting situation remains limited to comonomer pairs (i.e. the copolymerization of several different xx and yy comonomers leads to statistical sequences). Nevertheless, it has been demonstrated that is possible to create ABC ordered sequences in a step-growth polymerization using sequential approaches or monomer with selected reactivities.21 Nevertheless, these interesting strategies remain, on the whole, rather limited.
Yet, step-growth polymerizations can be easily transformed into sequence-specific oligomerizations if appropriate protection-deprotection cycles are performed. For instance, reactive monomers of the xy-type can be sequentially polymerized if one of two functions is momentarily deactivated (Scheme 1, bottom left). In this approach, an undefined batch polymerization is simply decomposed into discrete reaction-deprotection-isolation steps. Moreover, this process is greatly simplified if the growing oligomer is attached to a support (i.e. crosslinked polymer beads or high molecular weight polymer chains) allowing straightforward and rapid purification steps. Such supported-synthesis was first optimized for the sequence-ordered oligomerization of α-amino acids.22 In this approach, the C-terminus of a N-protected amino acid is first linked to a crosslinked poly(styrene-co-divinyl benzene) bead via a labile covalent linkage. After attachment, the N-terminus of this first amino acid is deprotected and reacted with the C-terminus of a second N-protected amino acid, in the presence of an ester activator. Such deprotection/amidification cycles can be repeated several times to access oligomers with defined monomer sequences. Solid-phase supported sequential oligomerization has been principally applied for the synthesis of sequence-defined biopolymers such as oligopeptides, oligonucleotides and oligosaccharides.17 However, this strategy can be theoretically extended to any kind of monomer of the xy-type if one of the two reactive functions is temporarily protected. For instance, solid-phase synthesis has been explored for preparing new types of sequence-defined macromolecules such as oligoureas, oligocarbamates, oligoesters, polyamides and polyamidoamines.23–27 However, these approaches remain restricted to short oligomer synthesis.
2.2 Chain-growth polymerizations
Controlling primary structure in a chain-growth polymerization (i.e. polymerizations consisting of chain-initiation and chain-propagation steps) is, theoretically speaking, much more challenging than in a step-growth process.28 Indeed, propagation steps rely on highly reactive transient species (e.g. radicals or ions), which are difficult to tame. Thus, the chain-growth copolymerization of two monomers A and B leads in most cases to undefined comonomer sequences (Scheme 1, top right). Depending on the intrinsic chemical reactivity of the comonomers (i.e. parameters which are quantified by the reactivity ratios or by the Q–e scheme, where Q expresses the monomer reactivity and e its polarization), statistical, random, block or alternating sequences can be theoretically obtained in a chain-growth radical, anionic or cationic polymerization (Scheme 1). The alternating case is particularly fascinating as it is one of the rare examples of perfectly controlled primary structures synthesized in a chain-growth process.29–33 Several alternating comonomers pairs have been described in the past. For example, the radical copolymerization of styrene (Q = 0.86; e = 3.69) and maleic anhydride (Q = 1; e = −0.8) is a classic example of an alternating process. However, although very interesting, chain-growth alternating polymerizations only lead to polymers containing repeating AB comonomer motifs. More advanced sequences have been prepared via a chain-growth mechanism.29 For instance, the terpolymerization of tetrahydrofuran, epichlorohydrin and phthalic anhydride leads to polymers containing predominant ABC sequences.34 However, such examples are unfortunately extremely rare.It should be noted that the chain-to chain sequence distribution of polymers prepared in a chain-growth polymerization is also strongly dependent on the reaction mechanism. For example, a distinct comonomer pair does not always lead to the same microstructures in a conventional free radical polymerization (i.e. a non-living polymerization) and in a controlled/“living” radical polymerization process such as atom transfer radical polymerization (ATRP), nitroxide mediated polymerization (NMP) or reversible addition-fragmentation chain transfer polymerization (RAFT).35–40 For example, the free radical polymerization of comonomer pairs with pronounced differences in reactivities usually leads to polymer with a heterogeneous chain-to-chain composition, while the ATRP of the same comonomers generates gradient copolymers with a homogeneous chain-to-chain composition.41,42 Similarly, alternating comonomer pairs may lead to different situations in living or non-living radical processes.43 Besides reaction mechanisms, the sequence distribution in radical polymerizations can be influenced by technical protocols. For example, the controlled addition (i.e. semi-batch mode) of one comonomer can lead to the formation of forced gradient microstructures.41
2.3 Polymer modification
Some examples of ordered primary structures obtained via polymer modification have been described in the literature. For example Nishikubo and coworkers reported the regioselective insertion of thiirane motifs into poly(s-aryl thioester) chains.441H NMR and 13C NMR spectroscopy indicated that this original method effectively led to the formation of sequence-ordered macromolecules. Yet, although very elegant, these kinds of methods remain marginal and limited to very particular types of polymers.3. Perspectives
As seen in the previous section, comonomer sequence distributions, obtained in standard chain-growth and step-growth polymerization processes, are nowadays well understood and accurately characterized. For instance, analytical methods such as high resolution NMR and MALDI-TOF mass spectrometry allow a relatively precise examination of polymer microstructures.45,46 However, “understanding” and “controlling” are two very different things. Even though the limitations of the present situation are obvious, concrete options for controlling the primary structure of synthetic polymers are still missing. In fact, our current state of knowledge in the field of macromolecular sequences is more or less the same as twenty years ago. Presently, defined synthetic sequences ABCDEF can only be prepared on a solid support, i.e. an approach which remains limited to the synthesis of short oligomers. Thus, alternative strategies have to be urgently developed. For instance, the sub-sections below discuss some possible directions for controlling macromolecular sequences. This is, of course, only a sample of what could be done. Several other chemical, physicochemical or even biochemical approaches may be considered. However, some of the methods described below have been already studied and therefore constitute an interesting starting point for future research in the field of macromolecular sequences.3.1 Templating
Macromolecular templating is an obvious strategy for controlling comonomer sequences. Indeed, sequence-specific biological polymerizations such as DNA replication, transcription and translation (i.e. protein synthesis) all rely on a template mechanism.14,47 However, templated polymerizations have been shown in the past to be rather disappointing for controlling comonomer sequences in synthetic processes. For instance, several research groups have investigated in vitro simplifications of biological templating for synthesizing sequence-controlled unnatural nucleic acids.48–50 In most cases, it was found that the sequence specificity of these templated oligomerizations is very limited. The overall yields of formed polymers with correct sequence information rarely exceeded a few percent. The point is that these in vitro versions of biological processes are too reductive. In synthetic macromolecular science, a template mechanism simply denotes a polymerization performed in the presence of a complementary macromolecule.51,52 In such processes, multiple monomer molecules can be simultaneously associated with the macromolecular template. In addition, an equilibrium of association/dissociation between the template and the monomers has to be considered.51 In biological polymerizations, these aspects are considerably more controlled. Indeed, biological monomers are usually associated one by one to their template and therefore polymerized in an exact sequential order. This precise monomer selection is also due to the presence of complex biocatalysts such as polymerases or ribosomes. Thus, in our present state of knowledge, it seems difficult and premature to recreate such complex mechanisms in a chemical process. Nevertheless, a very elegant model system was very recently investigated by Sawamoto and coworkers.53 This approach combines a template strategy with a controlled monomer insertion process. This promising example is discussed in detail in section 3.4.Yet, from a more philosophical point of view, it is questionable whether a macromolecular template is really necessary in a manmade synthetic experiment. Indeed, in biological polymerizations, the sequence information needs to be present in the form of a prewritten macromolecular template because these reactions indeed proceed in the absence of a guiding intelligence. In contrast, synthetic methods do not necessarily need inside information as sequences can be controlled by an external will (i.e. the sequence information is prewritten in the brain of the experimenter or in the memory of a computer). For example, Merrifield synthesis is a convincing example of efficient “template-free” sequence-controlled approach. Thus, it is important to specify that sequence-defined synthetic processes do not necessarily need to be bio-inspired to be successful.
3.2 Kinetic control
Polymerization kinetics have a marked influence on macromolecular sequences. For instance, as summarized in section 2.2, the rates of homopolymerization and copolymerization of a given comonomer pair are primarily determining the microstructure of the formed polymer. Thus, in some particular cases (e.g. alternating comonomer pairs), polymers with perfectly controlled comonomer sequences can be synthesized. However, as mentioned previously, these defined primary structures contain at most three different comonomers. Yet, these interesting kinetic situations can be adapted for synthesizing more advanced macromolecular sequences. For instance, we recently reported a simple method for controlling comonomer sequence distributions in a radical chain-growth polymerization.54,55 This concept relies on the atom transfer radical copolymerization of functional N-substituted maleimides with styrene (Fig. 1). This copolymerization is a controlled radical process, which combines two unique kinetic features: (i) all the polymer chains are growing simultaneously and (ii) the cross-propagation of the comonomers is highly favored as compared to homopolymerization (i.e. standard alternating scenario). Thus, discrete amounts of N-substituted maleimides (e.g. 1 eq. as compared to initiator) are consumed extremely rapidly in the copolymerization process and are therefore locally incorporated in narrow regions of the growing polystyrene chains.55 Typically, if a single equivalent of N-substituted maleimide is used in the copolymerization feed, short copolymer sections containing on average 1 or 2 functional maleimide units are preferentially formed. Furthermore, the position of the N-substituted maleimides in the polystyrene chains can be kinetically controlled by adding them at desired times during the course of the polymerization. This method is actually very versatile and can be applied to a wide variety of N-substituted maleimides. For instance, we screened in a previous article a library of 20 different maleimides bearing various functional groups (some examples are displayed in Fig. 1).55 In most cases, the functional N-substituted maleimides could be efficiently incorporated in the polystyrene chains. Moreover, this concept was utilized for preparing macromolecules with programmed sequences of functional comonomers. For example, we demonstrated that four different N-substituted maleimides could be consecutively added during the atom transfer radical polymerization of styrene.54 Indeed, the formed copolymers are not strictly sequence-defined at the molecular level (i.e. they still exhibit a sequence distribution). However, they undoubtedly possess a pre-programmed distribution of functional side-groups along the polymer backbone. Therefore, this straightforward copolymerization strategy can be used to prepare 1D functional arrays on linear polystyrene chains.56 However, these reactions remain challenging if one intends to create long sequences of functional comonomers. For instance, it seems difficult to kinetically control the addition of 5 maleimides or more without automated assistance (e.g. online monitoring of monomer conversions and automated maleimide additions).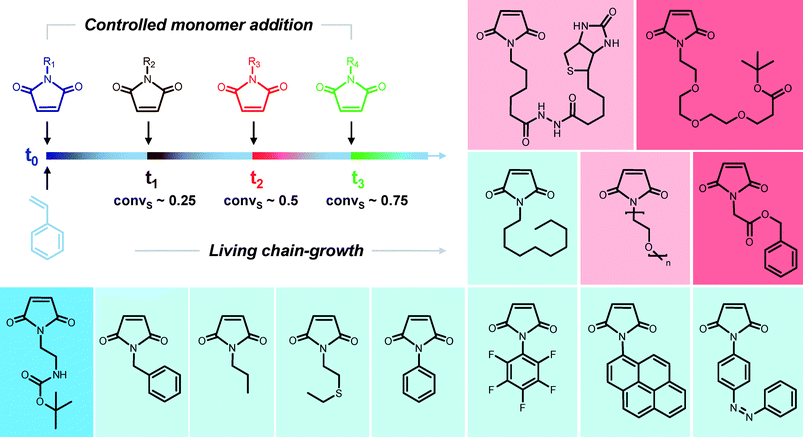 | ||
| Fig. 1 A concept for controlling sequences in a chain-growth polymerization: sequential atom transfer radical copolymerization of styrene and various N-substituted maleimides.54 The boxed structures show examples of N-substituted maleimides, which have been studied in this strategy.55 Color code: apolar monomers (light blue); monomers bearing a hydrophilic moiety (pink); monomers with a protected basic function (dark blue); monomers with a protected acidic function (red). | ||
3.3 Selected reactivities
It seems possible to regulate comonomer sequences in chain-growth polymerizations by selecting monomers or catalysts with very particular reactivities. A beautiful example was for example reported by Higashimura and coworkers. In a series of articles, these authors investigated an appealing concept for synthesizing sequence-ordered oligomers by living cationic polymerization.57,58 In this strategy, a combination of HI and ZnI2 was used to control the sequential oligomerization of vinyl ethers and styrene derivatives. For instance, HI reacts with a first monomer to form a stable iodo adduct. The latter is a dormant species, which is only activated by ZnI2. Hence, if this dormant adduct is activated in the presence of an equimolar equivalent of a second monomer, an iodo-terminated diadduct should be primarily formed. Theoretically, this strategy could be repeated to achieve sequence-defined oligomers. However, such an approach can only work if the activation/addition steps are kinetically favored as compared to homopolymerization. For instance, relatively monodisperse tetramers could be synthesized by reacting consecutively four monomers of decreasing reactivity.58Catalyst design is also a valid option toward sequence regulation. Very recently, Thomas and coworkers described an innovative approach for synthesizing biodegradable polyesters with a controlled primary structure.59 In this process, the monomer sequence was precisely controlled by a syndiospecific catalyst. In general, these types of catalysts are only used to synthesize syndiotactic homopolymers. However, in this work, the authors did not copolymerize a racemic mixture of a given monomer but two enantiopure comonomers with an opposite stereoconfiguration. As a result, unprecedented alternating polyesters have been synthesized in this process.
3.4 Controlled monomer insertion
The specific recognition and subsequent insertion of a given monomer unit would be an interesting pathway for controlling sequences in a synthetic polymerization process. Actually, all sequence-specific biological polymerizations utilize such a controlled insertion mechanism.47 However, current synthetic catalysts are certainly not evolved enough to play the same role as polymerases or ribosomes. Still, some interesting preliminary works have been described. For instance, Sawamoto and coworkers recently reported an elegant example of specific monomer insertion (Fig. 2).53 Generally speaking, the scientific objective of these authors is to use a template macroinitiator prepared by acid-catalyzed living cationic polymerization to regulate monomer insertion in a controlled/living radical polymerization process (Fig. 2, top). Such an ambitious goal certainly needs long-term development and therefore the authors studied at first a simplified low molecular weight model (Fig. 2, bottom). In this case, a dual initiator for both living cationic and living radical polymerizations was first designed (structure 1 in Fig. 2). Subsequently, the “cationic” site of this molecule was used to initiate a single monomer addition of a vinyl ether containing a pendant primary amine function. It was then demonstrated that the formed template (structure 2 in Fig. 2) allows the precise molecular recognition and radical incorporation of a single methacrylic acid monomer unit. These preliminary results are indeed fascinating and open some interesting perspectives for the future development of sequence-specific radical polymerizations.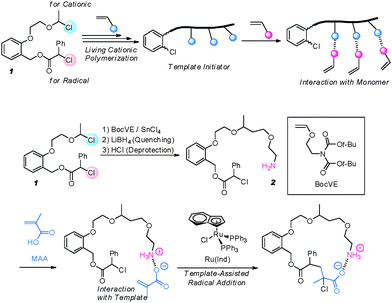 | ||
| Fig. 2 General concept for a sequence-regulated controlled/living radical polymerization in the presence of a template macroinitiator (top). Specific monomer insertion of methacrylic acid using a low molecular weight template initiator (bottom). Adapted with permission of the authors.53 Copyright 2009 American Chemical Society. | ||
3.5 Sequential growth on soluble polymer supports
As mentioned earlier, Merrifield solid-phase synthesis remains the most suitable route for preparing sequence-ordered macromolecules. This method is certainly efficient for synthesizing short oligomers but somewhat tedious for the preparation of complex macromolecular architectures. For instance, block copolymers containing sequence-defined segments are typically obtained by (i) synthesizing an oligomer on a solid support, (ii) cleavage and isolation of the oligomer and (iii) linking the oligomer with another polymer in solution (i.e. using coupling or macroinitiator strategies).17 The latter step could certainly be bypassed if the sequence-ordered oligomers are directly grown on a soluble polymer segment. Indeed, linear macromolecules can be easily isolated from low molecular weight mixtures (e.g. via selective precipitation) and therefore used as efficient supports for organic synthesis.60–63 Such soluble polymer supports interestingly combine the advantages of solid-phase synthesis (i.e. facile isolation) and solution chemistry (i.e. accessibility). For instance, some examples of oligonucleotide and oligopeptide synthesis on soluble polymer supports have been described in past years.64–66 However, in these approaches, the soluble polymers have been principally used as sacrificial supports. Thus, in most cases, ill-defined commercial polymers have been exploited. Yet, modern polymer chemistry certainly allows the design of more advanced macromolecular supports. For instance, controlled radical polymerization techniques such as ATRP and RAFT open wide possibilities in terms of macromolecular engineering.67–72 Nevertheless, these techniques have been barely explored to date for synthesizing tailored polymer supports.73In this context, we recently studied the synthesis of sequence defined oligomers on tailor-made soluble polymer supports.74 Monodisperse sequence-defined oligomers have been synthesized in solution and in the absence of protecting groups. These structures have been prepared stepwise using two consecutive chemoselective reactions: the 1,3 dipolar cycloaddition of terminal alkynes and azides and the amidification of carboxylic acids with primary amines. These oligomers were efficiently constructed either on a conventional solid support (commercial Wang resin) or on tailor-made soluble polystyrene supports synthesized by atom transfer radical polymerization (Fig. 3). The latter approach was found to be very versatile. Indeed, well-defined soluble macromolecular supports allowed the synthesis and cleavage of defined oligomers (i.e. sacrificial support) but also the preparation of non-cleavable block copolymers containing sequence-defined segments. This approach could be broadened to synthesize a wide variety of macromolecular architectures (e.g. block, star or graft topologies) containing sequence-ordered motifs.
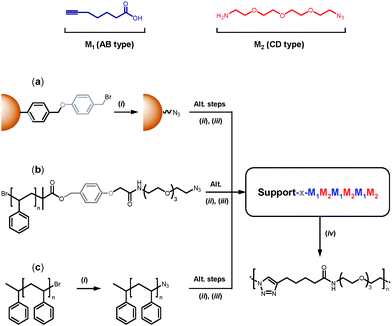 | ||
| Fig. 3 Strategies for synthesizing sequence-defined oligomers via an AB + CD growth mechanism (the capital letters A, B, C and D do not represent monomer units but reactive chemical functions): (a) conventional solid-phase approach based on an azido-functionalized Wang resin; (b) liquid-phase approach based on a linear polystyrene support containing a cleavable linker; (c) direct synthesis of block copolymers using an ω-azido functionalized polystyrene support. Reprinted with permission from ref. 74. Copyright 2009 American Chemical Society. | ||
3.6 Reconstitution
Most of the perspectives described in the above sectionss remain limited to the synthesis of relatively low molecular weight oligomers. Currently, it seems difficult to foresee sequence-regulated synthetic processes allowing the direct synthesis of high molecular weight macromolecules. However, the latter could be synthesized by linking together short sequence-defined oligomers. Such “patchwork” or “reconstitution” approaches are for example used nowadays for the total chemical synthesis of proteins.75 For instance, native chemical ligation has been shown to be a reliable technique for assembling short sequence-ordered oligopeptides into high molecular weight proteins.76 Similar strategies can be certainly applied in synthetic polymer science. For instance, the combination of efficient ligation techniques (e.g. macromolecular click coupling) with strategic convergent steps could allow the design of unprecedented high molecular weight sequence-ordered macromolecules.77–81 For example, the exponential-growth strategy recently described by Hawker and coworkers could be an interesting platform for controlling monomer sequences.82,83 Indeed, this approach was shown to afford monodisperse linear polymers with relatively high molecular weights (i.e. ∼5000 g mol−1).Alternatively, high molecular weight polymers could be also obtained via periodic polymerization of short sequence-defined motifs. For example, ring opening polymerizations and step-growth polymerizations have been both explored for the synthesis of periodic copolymers.85,86 For instance, the polymerization of short oligopeptides containing 3 to 10 ordered monomer units has been already reported in the literature.87–89 For example, Guan and coworkers investigated recently the step-growth “click” polymerization of short sequence-defined peptides (Fig. 4).84 This strategy allowed the straightforward design of high molecular weight macromolecules with precisely controlled primary and secondary structures. Our group also studied recently a simple step-growth strategy for synthesizing high molecular weight polymers containing periodic functional motifs.56 In this approach, short polystyrene segments containing functional units (e.g. benzyl, FmocNH- or tert-butyl ester) in the middle of their chains have been first synthesized using the maleimide strategy described in section 3.2. Subsequently, these well-defined oligomers have been transformed into reactive α-alkyne, ω-azido heterotelechelic polymers. These reactive precursors were ultimately polymerized by step-growth “click” coupling.90 SEC and 1H NMR measurements evidenced that high molecular weight periodic copolymers were formed in all cases. Thus, this approach appears as a straightforward method for preparing 1D molecular arrays.
![Concept of cycloaddition-induced folding and self-assembly: (a) [2 + 3] cycloaddition polymerization of a protected sequence-defined peptide monomer; (b) upon deprotection polypeptides fold into well-defined antiparallel β-strands; (c) self-assembly of multiple β-sheets forms hierarchical nanofibrils. Adapted with permission from ref. 84. Copyright 2009 Wiley-VCH.](/image/article/2010/PY/b9py00329k/b9py00329k-f4.gif) | ||
| Fig. 4 Concept of cycloaddition-induced folding and self-assembly: (a) [2 + 3] cycloaddition polymerization of a protected sequence-defined peptide monomer; (b) upon deprotection polypeptides fold into well-defined antiparallel β-strands; (c) self-assembly of multiple β-sheets forms hierarchical nanofibrils. Adapted with permission from ref. 84. Copyright 2009 Wiley-VCH. | ||
4. Outlook
The control over polymer architecture has been one of the biggest achievements of polymer chemists during the last two decades. Realistically speaking, the synthesis of well-defined polymers with controlled primary structures could be the next breakthrough in polymer design. However, the current situation is not really propitious for such a revolution. Indeed, although very interesting concepts have been recently reported, the efforts in this crucial area of research are still way too little. Nevertheless, some examples discussed in the present manuscript clearly indicate that the goal is reachable.Interestingly, one of the referee of this manuscript partially disagreed with the previous sentence. He/she argued that no statistical process can provide a perfect sequence control. This is of course very true. However, the views described in this preceding section should not be overinterpreted. It is clear that realistic scientific objectives have to be set for the short and medium term. For instance, in standard statistical polymerizations (i.e. chain-growth or step-growth), a reasonable goal would be to narrow down comonomer sequence distributions. Within the last years, several pathways for controlling chain-length distributions in statistical processes have been discovered. Similarly, strategies for taming comonomer sequence distributions could be envisaged. The styrene/N-substituted maleimide copolymerization approach, described in section 3.2, is one example in that direction.
Still, for the longer term, monodisperse synthetic macromolecules with controlled primary structures may be envisioned. Indeed, in these cases, alternative polymerization tools have to be imagined. For example, some of the options described in section 3.6 could be interesting starting points. Nevertheless, it is certain that several decades of research are needed to attain that ultimate goal, but hopefully the student textbooks of the 22nd century will not describe statistical comonomer sequences as the state-of-the-art in polymer science.
Acknowledgements
The Fraunhofer Society and the Federal Ministry of Education and Research are acknowledged for financial support. Additionally, J.F.L. thanks Professor André Laschewsky (Universität Potsdam) for fruitful discussions and Professor Dave Haddleton (University of Warwick) for his kind invitation to write the present manuscript.Notes and references
- P. Mansky, Y. Liu, E. Huang, T. P. Russell and C. Hawker, Science, 1997, 275, 1458–1460 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- Z. Li, E. Kesselman, Y. Talmon, M. Hillmyer and T. Lodge, Science, 2004, 306, 98–101 CrossRef CAS.
- A. Harada and K. Kataoka, Science, 1999, 283, 65–67 CrossRef CAS.
- F. S. Bates, Science, 1991, 251, 898–905 CAS.
- S. Förster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688–714 CrossRef CAS.
- I. W. Hamley, Angew. Chem., Int. Ed., 2003, 42, 1692–1712 CrossRef CAS.
- S. Kubowicz, J.-F. Baussard, J.-F. Lutz, A. F. Thünemann, H. von Berlepsch and A. Laschewsky, Angew. Chem., Int. Ed., 2005, 44, 5262–5265 CrossRef CAS.
- M. Arotçaréna, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787–3793 CrossRef CAS.
- J. Z. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- J.-F. Lutz, S. Geffroy, H. von Berlepsch, C. Böttcher, S. Garnier and A. Laschewsky, Soft Matter, 2007, 3, 694–698 RSC.
- J.-F. Lutz, Polym. Int., 2006, 55, 979–993 CrossRef CAS.
- R. Jones, Nat. Nanotechnol., 2008, 3, 699–700 CrossRef CAS.
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- J. J. Storhoff and C. A. Mirkin, Chem. Rev., 1999, 99, 1849–1862 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- J.-F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- H. G. Börner and H. Schlaad, Soft Matter, 2007, 3, 394–408 RSC.
- M. Safak, F. E. Alemdaroglu, Y. Li, E. Ergen and A. Herrmann, Adv. Mater., 2007, 19, 1499–1505 CrossRef CAS.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- M. Ueda, Prog. Polym. Sci., 1999, 24, 699–730 CrossRef CAS.
- R. B. Merrifield, Angew. Chem., Int. Ed. Engl., 1985, 24, 799–810 CrossRef.
- C. Cho, E. Moran, S. Cherry, J. Stephans, S. Fodor, C. Adams, A. Sundaram, J. Jacobs and P. Schultz, Science, 1993, 261, 1303–1305 CrossRef CAS.
- K. Burgess, H. Shin and D. S. Linthicum, Angew. Chem., Int. Ed. Engl., 1995, 34, 907–909 CrossRef CAS.
- K. Rose and J. Vizzavona, J. Am. Chem. Soc., 1999, 121, 7034–7038 CrossRef CAS.
- T. M. Fyles, C. W. Hu and H. Luong, J. Org. Chem., 2006, 71, 8545–8551 CrossRef CAS.
- L. Hartmann and H. G. Börner, Adv. Mater., 2009, 21, 3425–3431 CrossRef CAS.
- J.-F. Lutz, T. Pakula and K. Matyjaszewski, ACS Symp. Ser., 2003, 854, 268–282 CAS.
- T. Saegusa, Makromol. Chem., 1979, 3, 157–176 CrossRef CAS.
- J. M. G. Cowie, Alternating Copolymers, Plenum Press, New York, 1985 Search PubMed.
- Z. M. O. Rzaev, Prog. Polym. Sci., 2000, 25, 163 CrossRef CAS.
- B. Kirci, J.-F. Lutz and K. Matyjaszewski, Macromolecules, 2002, 35, 2448–2451 CrossRef CAS.
- J.-F. Lutz, B. Kirci and K. Matyjaszewski, Macromolecules, 2003, 36, 3136–3145 CrossRef CAS.
- H. L. Hsieh, US Pat., 3661865, 1972.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- K. Matyjaszewski, M. J. Ziegler, S. V. Arehart, D. Greszta and T. Pakula, J. Phys. Org. Chem., 2000, 13, 775–786 CrossRef.
- J.-F. Lutz, N. Jahed and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1939–1952 CrossRef CAS.
- D. Benoit, C. J. Hawker, E. E. Huang, Z. Lin and T. P. Russell, Macromolecules, 2000, 33, 1505–1507 CrossRef CAS.
- A. Kameyama, Y. Murakami and T. Nishikubo, Macromolecules, 1996, 29, 6676–6678 CrossRef CAS.
- F. A. Bovey and P. A. Mirau, NMR of polymers, Academic Press, Inc., San Diego, 1996 Search PubMed.
- G. Montaudo and R. P. Lattimer, ed., Mass Spectrometry of Polymers, CRC Press LLC, Boca Raton, 2001 Search PubMed.
- J. M. Berg, Tymoczko and L. Stryer, Biochemistry, 6th edn, W.H. Freeman, New York, 2006 Search PubMed.
- L. D. Kosturko, N. Dattagupta and D. M. Crothers, Biochemistry, 1979, 18, 5751–5756 CrossRef CAS.
- L. E. Orgel, Acc. Chem. Res., 1995, 28, 109–118 CrossRef CAS.
- X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS.
- Y. Y. Tan, Prog. Polym. Sci., 1994, 19, 561–588 CrossRef CAS.
- S. Polowinski, Prog. Polym. Sci., 2002, 27, 537–577 CrossRef CAS.
- S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808–10809 CrossRef CAS.
- S. Pfeifer and J.-F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS.
- S. Pfeifer and J.-F. Lutz, Chem.–Eur. J., 2008, 14, 10949–10957 CrossRef CAS.
- M. A. Berthet, Z. Zarafshani, S. Pfeifer and J.-F. Lutz, Macromolecules DOI:10.1021/ma902075q.
- M. Minoda, M. Sawamoto and T. Higashimura, Polym. Bull., 1990, 23, 133–139 CrossRef CAS.
- M. Minoda, M. Sawamoto and T. Higashimura, Macromolecules, 1990, 23, 4889–4895 CrossRef CAS.
- (a) J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042–16044 CrossRef CAS; (b) J.-F. Lutz, Nat. Chem. DOI:10.1038/nchem.530.
- D. J. Gravert and K. D. Janda, Chem. Rev., 1997, 97, 489–510 CrossRef CAS.
- P. Wentworth Jr. and K. D. Janda, Chem. Commun., 1999, 1917–1924 RSC.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS.
- S. Pfeifer and J.-F. Lutz, Macromol. Chem. Phys. DOI: 10.1002/macp.200900678.
- M. M. Shemyakin, Y. A. Ovchinnikov, A. A. Kinyushkin and I. V. Kozhevnikova, Tetrahedron Lett., 1965, 6, 2323–2327 CrossRef.
- H. Hayatsu and H. G. Khorana, J. Am. Chem. Soc., 1966, 88, 3182–3183 CrossRef CAS.
- E. Bayer and M. Mutter, Nature, 1972, 237, 512–513 CAS.
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS.
- K. V. Bernaerts and F. E. Du Prez, Prog. Polym. Sci., 2006, 31, 671–722 CrossRef CAS.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 Search PubMed.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- D. J. Gravert, A. Datta, P. Wentworth and K. D. Janda, J. Am. Chem. Soc., 1998, 120, 9481–9495 CrossRef CAS.
- S. Pfeifer, Z. Zarafshani, N. Badi and J.-F. Lutz, J. Am. Chem. Soc., 2009, 131, 9195–9197 CrossRef CAS.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS.
- S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS.
- I. Singh, Z. Zarafshani, J.-F. Lutz and F. Heaney, Macromolecules, 2009, 42, 5411–5413 CrossRef CAS.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- K. Takizawa, H. Nulwala, J. Hu, K. Yoshinaga and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5977–5990 CrossRef CAS.
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718–1726 CrossRef CAS.
- T. B. Yu, J. Z. Bai and Z. B. Guan, Angew. Chem., Int. Ed., 2009, 48, 1097–1101 CrossRef CAS.
- K. Yokota, Prog. Polym. Sci., 1999, 24, 517–563 CrossRef CAS.
- I. Cho, Prog. Polym. Sci., 2000, 25, 1043–1087 CrossRef CAS.
- D. W. Urry, T. L. Trapane and K. U. Prasad, Biopolymers, 1985, 24, 2345–2356 CrossRef CAS.
- D. W. Urry, J. Phys. Chem. B, 1997, 101, 11007–11028 CrossRef CAS.
- Y. Tachibana, N. Matsubara, F. Nakajima, T. Tsuda, S. Tsuda, K. Monde and S.-I. Nishimura, Tetrahedron, 2002, 58, 10213–10224 CrossRef CAS.
- N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2005, 38, 3558–3561 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |