Highly permeable polymers for gas separation membranes
Peter M.
Budd
*a and
Neil B.
McKeown
b
aSchool of Chemistry, The University of Manchester, Manchester, M13 9PL, UK. E-mail: Peter.Budd@manchester.ac.uk; Fax: +44 1612754273; Tel: +44 1612754711
bSchool of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: McKeownnb@cardiff.ac.uk; Fax: +44 2920874030; Tel: +44 2920875851
First published on 4th January 2010
Abstract
For gas separation membranes, materials are required that offer high permeability as well as good selectivity for a desired separation. For glassy polymers, the gas transport properties depend on the amount and distribution of free volume and on chain mobility. The most highly permeable polymers have rigid, twisted macromolecular backbones that give rise to microvoids. Examples include substituted polyacetylenes, perfluoropolymers, addition-type polynorbornene, polymers of intrinsic microporosity (PIMs) and some polyimides. High permeability membranes may also be produced by thermal rearrangement of precursor polymers.
Introduction
Gas separation membranes are increasingly being used for applications such as hydrogen recovery, nitrogen generation and carbon dioxide removal.1 Membrane technology is seen as a route to cost-effective, energy-efficient, environmentally benign processes in many industries. However, research is still needed to generate better and more robust membrane materials, capable of maintaining their properties for a long period under the demanding conditions encountered in practice.Many polymers have been investigated as gas separation membrane materials, but up to now only a handful have found commercial success. These include rubbery polymers, such as poly(dimethylsiloxane), and glassy polymers, such as polysulfone, poly(phenylene oxide), cellulose acetate and polyimides. To be useful, a membrane material must exhibit good selectivity for a particular species in a mixture, in order to achieve sufficient separation, as well as good permeability, to minimize the membrane area required. Unfortunately, polymers that exhibit good selectivity generally have low permeability and vice versa. This trade-off behaviour can be depicted for a pair of gases by a double logarithmic plot of selectivity (expressed as a ratio of permeabilities) against the permeability of the fastest species (Fig. 1). Robeson demonstrated an empirical upper bound in such plots in 19912 and updated the upper bounds in 2008.3 Note that these trade-off plots refer to ideal behaviour and do not take account of plasticization or other effects. Theories of the trade-off behaviour have been developed.4–6
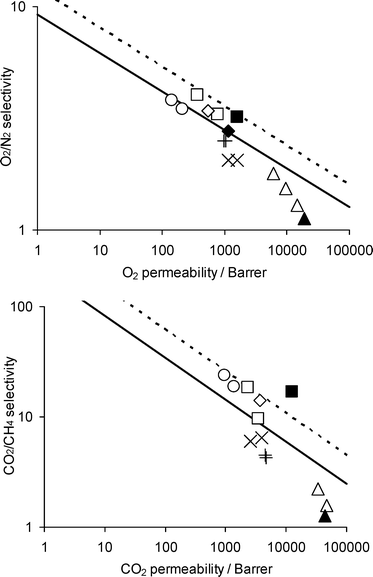 | ||
| Fig. 1 Double logarithmic plots of selectivity versus permeability for (top) O2/N2 and (bottom) CO2/CH4, showing (solid line) Robeson's 1991 upper bound2 and (dashed line) 2008 upper bound,3 and data for (△) poly(trimethylsilyl propyne) (PTMSP),7–9 (▲) indan-based polyacetylene 2e,9 (×) Teflon AF2400,10,11 (+) addition-type poly(trimethylsilyl norbornene),12,13 (□) PIM-1,14,15 (■) PIM-1 after methanol treatment,16 (○) 6FDA-DMN polyimide,17,18 (◇) PIM-PI-8,19 and (♦) PIM-PI-8 after methanol treatment.20 | ||
Glassy polymers are most commonly employed for membrane gas separations, because they are capable of high selectivity and have good mechanical properties. The challenge for polymer chemists is to develop polymers with much higher permeability, whilst retaining adequate selectivity and meeting other requirements, such as processability and long-term stability. This review discusses recent work on highly permeable glassy polymers. First, some essential background is provided on membrane gas separation and on free volume in polymers.
Membrane gas separation
The membrane gas separation process is driven by a pressure difference across the membrane. The membrane may be in the form of a flat sheet or a hollow fibre, the latter being generally preferred because a much higher effective membrane area is achievable within a given module volume. A single module may contain thousands of fibres.The productivity of a membrane is expressed in terms of its permeance, a measure of the amount of permeate that passes through a certain membrane area in a given time for a particular pressure difference. Permeance multiplied by thickness gives the permeability P (more strictly called the permeability coefficient), which is a characteristic of the material. In principle, permeability is independent of membrane thickness for a homogeneous membrane, but in practice values can depend both on the thickness of the membrane and on its history. Values of permeability are often quoted in units of Barrer (1 Barrer = 10−10 cm3 [STP] cm cm−2 s−1 cmHg−1 = 3.35 × 10−16 mol m m−2 s−1 Pa−1). This review is concerned with polymers that exhibit an oxygen permeability greater than 100 Barrer.
Initial permeability measurements are frequently made on relatively thick (20–100 μm) homogeneous membranes, but for practical application very thin active layers (<1 μm) are desired in order to increase the permeance. For suitable polymers, a so-called phase inversion process may be used to generate asymmetric membranes with a thin, dense surface layer and a highly porous sub-layer. Alternatively, a thin separating layer may be coated on to a porous substrate, giving a composite membrane.
The simplest model of permeation involves (i) dissolution (or sorption) of the permeating species in the membrane at the high pressure side, (ii) diffusion through the membrane and (iii) dissolution (or desorption) at the low pressure side. In the simple solution-diffusion model, permeability is the product of solubility coefficient, S, and mutual diffusion coefficient, D:
| P = SD | (1) |
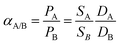 | (2) |
In the simplest case, the solubility of a gas is given by Henry's law, so that the concentration, C, of gas in a polymer is directly proportional to its partial pressure, p:
| C = kDp | (3) |
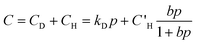 | (4) |
The uptake and transport of gases in a glassy polymer is linked to the amount and distribution of free volume, so this will be discussed next.
Free volume
The free volume in a polymer sample is the space not occupied by polymer molecules. The occupied volume is generally taken to include the van der Waals volume multiplied by a factor (typically 1.3) to take account of the fact that, even for a perfect crystal at absolute zero, there is a limit to the packing density achievable.21 On this basis, fractional free volume, fV, can be calculated as: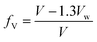 | (5) |
The glass transition may be thought of as occurring when there is sufficient fractional free volume for large scale polymer motions to occur. In the rubbery state, free volume increases markedly with increasing temperature. If a polymer is quenched from the rubbery into the glassy state, excess free volume is trapped. Over time, excess free volume may be lost, a process referred to as physical ageing. This can be particularly pronounced in very thin films, as used for gas separation.22,23 Long-term ageing has a profound influence on transport properties and represents a major problem in the application of glassy polymer membranes.
For high permeability we need high free volume. If there is so much free volume in a glassy polymer that the free volume elements are essentially interconnected, the polymer may exhibit adsorption behavour like that of a molecular sieve.24 In other words, it contains micropores (pores with dimensions <2 nm), as defined by IUPAC in the context of adsorption studies.25 It should be noted that in the membrane literature the word “microporous” is also often applied to materials with much larger pores (macroporous in the IUPAC sense). Ilinitch et al.26 argued on the basis of nitrogen adsorption studies that certain polyphenylene oxides possess interconnected microcavities and they referred to this as “intrinsic microporosity”. However, in polyphenylene oxides the level of microporosity is relatively low. For very high permeability, greater free volume and a better developed microporosity is required. The following sections indicate ways of achieving this.
High free volume polymers
The main types of high free volume glassy polymer are discussed below. Chemical structures and molecular models of representative examples are shown in Fig. 2 and Fig. 3, respectively. Membrane permeability and selectivity data for these polymers are plotted in Fig. 1 for the O2/N2 and CO2/CH4 gas pairs.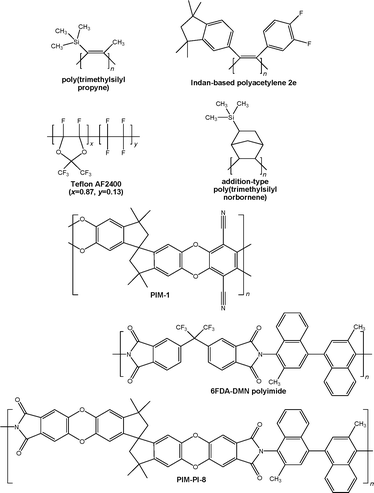 | ||
| Fig. 2 Chemical structures of important high free volume polymers. | ||
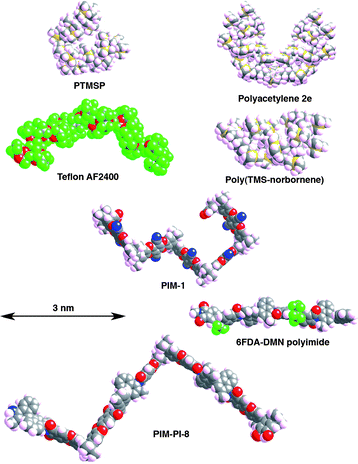 | ||
| Fig. 3 Molecular models of fragments of high free volume polymers (chemical structures shown in Fig. 2), illustrating different ways of creating an inflexible backbone. Note that more sophisticated simulations of packed macromolecules are required in order to predict free volume distribution and permeation properties. | ||
Substituted polyacetylenes
In 1983, Masuda et al.7 reported that a substituted polyacetylene, poly(1-trimethylsilyl-1-propyne) (PTMSP) exhibited an oxygen permeability ten times higher than that of the most permeable polymer then known, the rubbery polymer poly(dimethylsiloxane). PTMSP, although glassy, showed some properties that were similar to rubbers, including solubility-controlled permeation. This spawned a great deal of research into substituted polyacetylenes.27,28 Other highly permeable polyacetylenes were found, including poly(4-methyl pentyne) (PMP)29 and poly[1-phenyl-2-(p-trimethylsilylphenyl)acetylene].30 However, PTMSP remained the champion until 2008, when certain indan-containing poly(diphenylacetylene) derivatives were shown to exhibit even higher oxygen permeabilities.9Substituted polyacetylenes are generally prepared by polymerization of the acetylenic monomer using transition metal catalysts.28 In a polyacetylene, cis- and trans-arrangements are possible at each double bond. Different catalysts may give polymers with different geometric structures and different properties. In the case of PTMSP, a NbCl5 catalyst gives a more cis-rich polymer than TaCl5.31,32
The common feature of highly permeable acetylene-based polymers is the presence of bulky side groups that inhibit conformational change and force the backbone into a twisted shape.33 When these rigid, randomly coiled macromolecules are packed in the solid state, the free volume is large and the free volume distribution includes both small disconnected elements, as in conventional glassy polymers, and larger continuous microvoids. This has been demonstrated by molecular modelling and positron annihilation lifetime spectroscopy (PALS).34,35 Large microvoids are expected to give rise to high diffusion coefficients compared to other glassy polymers.36 High free volume also leads to large values for the dual-mode sorption parameter, C′H, and to high apparent solubility coefficients.37 The combination of large D and large S gives exceptionally high P.
The high permeabilities of PTMSP and related polymers come at the cost of rather low selectivities, in accordance with the trade-off behaviour discussed above. The selectivity is controlled by the solubility selectivity,37 unlike conventional glassy polymers for which mobility selectivity generally dominates.
For PTMSP a very wide range of values of permeability have been reported by different groups.7–9 Furthermore, large ageing effects have been observed, with permeability decreasing over time.38 This may be attributed to physical relaxation and/or vapour sorption. The effects of ageing may be reversed by soaking the membrane in methanol.39 Crosslinking the membrane may help to reduce ageing effects in some cases.33
Research on substituted polyacetylenes has demonstrated that subtle changes in chemical structure can have a profound impact on free volume and hence on the gas transport properties. There is still scope for improvement of polymer properties through design of the macromolecular structure. However, a step-change in performance is most likely to be achieved through the judicious addition of a second component. For example, addition of non-porous inorganic fillers to stiff-chain glassy polymers can enhance permeability and selectivity.40–42 “Mixed matrix” membranes represent an active area of current research.43
Perfluoropolymers
There are a number of commercially available perfluoropolymers (Teflon AF, Hyflon AD, Cytop) that form amorphous, glassy films.44 Of these, the highest permeabilities are obtained for Teflon AF2400 and Teflon AF1600, which are copolymers of 2,2-bistrifluoromethyl-4,5-difluoro-1,3-dioxole and tetrafluoroethylene with mole fractions of the dioxole of 0.87 and 0.65, respectively.10,11Teflon AF polymers have much lower densities than polytetrafluoroethylene (ca. 1.7 as compared to 2.2 g cm−3), reflecting their high free volume. As with PTMSP, molecular modelling35 and PALS45 indicate the presence of large microvoids, although they may have less continuity than in PTMSP. The key molecular feature of Teflon AF is a high barrier to rotation between neighbouring dioxolane rings (ca. 60 kJ mol−1),46 coupled with weak interchain interactions, as is typical for fluoropolymers. This leads to low packing density (high free volume) and hence high permeability.
Poly(norbornene)s
Norbornene derivatives may be polymerised either by ring-opening metathesis polymerization (ROMP) or by addition polymerization at the double bond. Most ROMP polynorbornenes show modest permeabilities.47 However, addition-type poly(trimethylsilyl norbornene), prepared with nickel naphthenate as catalyst and methylaluminoxane as co-catalyst, exhibits high free volume and high permeability.12,13 As with other very high free volume polymers, there is evidence from PALS of large microvoids.12 As with PTMSP, it seems that the bulky trimethylsilyl group on the ring greatly restricts the freedom of the polymer to undergo conformational change.Polymers of intrinsic microporosity
The high free volume polymers discussed above possess side groups and/or incorporate ring structures that inhibit free rotation about single bonds in the polymer backbone. The next step in the design of high free volume polymers is to form a polymer without any single bonds in the backbone; for example, a ladder polymer with a backbone of fused rings. Such a polymer cannot undergo large scale conformational change without breaking bonds. However, ladder polymers are generally dense, intractable materials. It is necessary, therefore, also to incorporate “sites of contortion” that force the backbone to twist and turn erratically, creating polymer chains that cannot pack efficiently in the solid state. This basic concept has been used by the present authors to generate polymers referred to as “polymers of intrinsic microporosity” (PIMs).48,49 The concept developed out of work aimed at producing high surface area polymer networks incorporating catalytic centres,50–53 but subsequently it was found that soluble, membrane-forming PIMs could be prepared.54,55The archetypal soluble PIM, referred to as PIM-1, is prepared from 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane and 1,4-dicyanotetrafluorobenzene by a step polymerization involving a double aromatic nucleophilic substitution.55–57 The spiro-centre (a tetrahedral carbon atom that is part of two five-membered rings) acts as a “site of contortion”. PIM-1 is soluble in tetrahydrofuran, chloroform, dichloromethane and o-dichlorobenzene. In the solid state, it is a yellow, fluorescent, glassy polymer with a degradation temperature >350 °C by thermogravimetric analysis. There is no discernible glass transition below the degradation temperature. Evidence for microporosity (pores <2 nm, as defined by IUPAC)25 comes from nitrogen sorption studies.14 This is supported by molecular modelling58 and PALS.15,59,60
Initial gas permeation experiments showed PIM-1 membranes to exhibit permeabilities exceeded only by very high free volume polymers such as PTMSP and Teflon AF2400, coupled with selectivities above Robeson's2 1991 upper bound for important gas pairs such as O2/N2 and CO2/CH4 (Fig. 1).14 Subsequent studies showed that permeability could be substantially enhanced by a methanol treatment that helps flush out residual casting solvent and allows relaxation of chains (Fig. 1).16 The properties of PIM-1 membranes may be tuned by chemical modification, such as carboxylation.61
Following on from PIM-1, a variety of monomers have been incorporated into soluble PIMs, including novel spirobisindanes,62,63 bis(phenazyl) monomers,64 2,2′,3,3′-tetrahydroxy-1,1′-dinaphthyl,65 heptafluoro-p-tolylphenylsulfone,66 and disulfone-based monomers.67
Polyimides
Polyimides have been extensively investigated for gas separation membranes.68 They are commonly prepared by a step polymerization involving a cycloimidization reaction between a bis(carboxylic anhydride) and a diamine. Many polyimides exhibit good selectivity, but at the expense of modest permeability. A few, however, show high permeability, in particular those prepared from 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA) with diamines such as 2,3,5,6-tetramethyl-1,4-phenyldiamine (4MPDA)69–71 and 3,3′-dimethylnaphthidine (DMN),17,18,72 for which rotation about the imide linkage is very highly restricted.To create even more permeable polyimides, the PIM concept was applied. A dianhydride was prepared that incorporated a spiro-centre.19,20,73 This was then reacted with a range of diamines, including 4MPDA (polymer designated PIM-PI-1) and DMN (polymer designated PIM-PI-8).19,20 PIM-PI-8 exhibits the highest permeabilities so far reported for a polyimide. As with PIM-1, permeability is enhanced by a methanol treatment (Fig. 1). By varying the structure of the diamine used, and hence the flexibility of the polymer backbone, it is possible to move up and down Robeson's upper bound. A group contribution method enables transport properties to be predicted.20
Thermally-rearranged polymers
The polymers discussed above are soluble polymers that can be cast from solution to form a high free volume (effectively microporous), glassy polymer film. Many other polymer structures can be envisaged that would exhibit desirable membrane properties, but which cannot be prepared in soluble form. An alternative route to high free volume membranes is to use a soluble precursor to prepare the membrane, and subsequently to generate the desired structure by thermal treatment. Park et al.74 demonstrated this approach using soluble aromatic polyimides with –OH or –SH groups in the ortho-position, which rearrange irreversibly to insoluble, infusible polybenzoxazoles or polybenzothiazoles on heating at temperatures of 350–450 °C (Fig. 4). These thermally rearranged (TR) polymers exhibit performances well above Robeson's upper bound for the CO2/CH4 gas pair. The properties can be tuned by adding acidic dopants which interact with basic nitrogen atoms in the heterocyclic rings.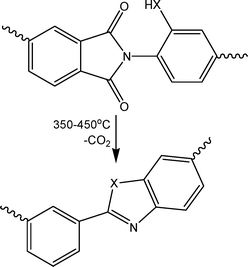 | ||
Fig. 4 Thermal rearrangement of polyimide to polybenzoxazole (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) or polybenzothiazole (XS). O) or polybenzothiazole (XS). | ||
Conclusions
In recent years, significant progress has been made in the development of glassy, high free volume polymers for membrane applications. These polymers have twisted backbones that frustrate packing and that cannot undergo large-scale conformational change, either because rotation about single bonds in the backbone is restricted due to bulky side groups, or because the backbone is made up of fused rings. However, the complex inter-relationships between molecular structure, free volume distribution and transport properties are not yet fully understood. Subtle changes in molecular architecture can have a profound effect on properties. It is not easy to predict whether a particular structural modification will fill space and reduce permeability or create space and enhance permeability. Predictions are complicated by the fact that glassy polymers are non-equilibrium. Thus, to control free volume distribution, and hence permeation behaviour, through design of the polymer structure represents a major intellectual challenge. Furthermore, the chemist working in this area needs to remain aware of the engineering and economic factors that are important if a material is to find real application. Although the cost of manufacture of the membrane system will be of prime importance, it should be noted that the cost of the highly permeable polymer is unlikely to contribute significantly due to the small amount used to form the sub-micron thick separation layer. Therefore, this is an area of technology where a more exotic polymer of higher performance may have a competitive advantage over a low-cost polymer of lesser performance.The goal of being able to design readily processible membrane materials that have desirable combinations of permeability and selectivity, with long-term stability, is challenging, but potentially highly rewarding. Success is likely to come about through a combination of clever polymer synthesis and subsequent processing to enhance performance and improve stability.
Notes and references
- P. Bernardo, E. Drioli and G. Golemme, Ind. Eng. Chem. Res., 2009, 48, 4638–4663 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS.
- A. Y. Alentiev and Y. P. Yampolskii, J. Membr. Sci., 2000, 165, 201–216 CrossRef CAS.
- M. M. Dal-Cin, A. Kumar and L. Layton, J. Membr. Sci., 2008, 323, 299–308 CrossRef CAS.
- T. Masuda, E. Isobe, T. Higashimura and K. Takada, J. Am. Chem. Soc., 1983, 105, 7473–7474 CrossRef CAS.
- I. Pinnau and L. G. Toy, J. Membr. Sci., 1996, 116, 199–209 CrossRef CAS.
- Y. Hu, M. Shiotsuki, F. Sanda, B. D. Freeman and T. Masuda, Macromolecules, 2008, 41, 8525–8532 CrossRef CAS.
- I. Pinnau and L. G. Toy, J. Membr. Sci., 1996, 109, 125–133 CrossRef CAS.
- A. Y. Alentiev, Y. P. Yampolskii, V. P. Shantarovich, S. M. Nemser and N. A. Plate, J. Membr. Sci., 1997, 126, 123–132 CrossRef CAS.
- E. S. Finkelshtein, K. L. Makovetskii, M. L. Gringolts, Y. V. Rogan, T. G. Golenko, L. E. Starannikova, Y. P. Yampolskii, V. P. Shantarovich and T. Suzuki, Macromolecules, 2006, 39, 7022–7029 CrossRef CAS.
- L. Starannikova, M. Pilipenko, N. Belov, Y. Yampolskii, M. Gringolts and E. Finkelshtein, J. Membr. Sci., 2008, 323, 134–143 CrossRef CAS.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS.
- C. L. Staiger, S. J. Pas, A. J. Hill and C. J. Cornelius, Chem. Mater., 2008, 20, 2606–2608 CrossRef CAS.
- P. M. Budd, N. B. McKeown, B. S. Ghanem, K. J. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. Yampolskii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851–860 CrossRef CAS.
- D. Hofman, J. Ulbrich, D. Fritsch and D. Paul, Polymer, 1996, 37, 4773–4785 CrossRef CAS.
- C. Nagel, K. Guenther-Schade, D. Fritsch, T. Strunskus and F. Faupel, Macromolecules, 2002, 35, 2071–2077 CrossRef CAS.
- B. S. Ghanem, N. B. McKeown, P. M. Budd, J. D. Selbie and D. Fritsch, Adv. Mater., 2008, 20, 2766–2771 CrossRef CAS.
- B. S. Ghanem, N. B. McKeown, P. M. Budd, N. M. Al-Harbi, D. Fritsch, K. Heinrich, L. Starannikova, A. Tokarev and Y. Yampolskii, Macromolecules, 2009, 42, 7881–7888 CrossRef CAS.
- A. Bondi, Physical Properties of Molecular Crystals, Liquids and Glasses, John Wiley & Sons, New York, 1968 Search PubMed.
- Y. Huang, X. Wang and D. R. Paul, J. Membr. Sci., 2006, 277, 219–229 CrossRef CAS.
- Y. Huang and D. R. Paul, Ind. Eng. Chem. Res., 2007, 46, 2342–2347 CrossRef CAS.
- P. M. Budd, N. B. McKeown and D. Fritsch, J. Mater. Chem., 2005, 15, 1977–1986 RSC.
- D. H. Everett, Pure Appl. Chem., 1972, 31, 577–638 CrossRef.
- O. M. Ilinitch, V. B. Fenelonov, A. A. Lapkin, L. G. Okkel, V. V. Terskikh and K. I. Zamaraev, Microporous Mesoporous Mater., 1999, 31, 97–110 CrossRef CAS.
- K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721–798 CrossRef CAS.
- T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2006, 45, 165–180.
- A. Morisato and I. Pinnau, J. Membr. Sci., 1996, 121, 243–250 CrossRef CAS.
- K. Tsuchihara, T. Masuda and T. Higashimura, J. Am. Chem. Soc., 1991, 113, 8548–8549 CrossRef CAS.
- H. Izumikawa, T. Masuda and T. Higashimura, Polym. Bull., 1991, 27, 193–199 CrossRef CAS.
- V. S. Khotimsky, M. V. Tchirkova, E. G. Litvinova, A. I. Rebrov and G. N. Bondarenko, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2133–2155 CrossRef CAS.
- J. Jia and G. L. Baker, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 959–968 CrossRef CAS.
- D. Hofmann, M. Heuchel, Y. Yampolskii, V. Khotimskii and V. Shantarovich, Macromolecules, 2002, 35, 2129–2140 CrossRef CAS.
- D. Hofmann, M. Entrialgo-Castano, A. Lerbret, M. Heuchel and Y. Yampolskii, Macromolecules, 2003, 36, 8528–8538 CrossRef CAS.
- X.-Y. Wang, R. D. Raharjo, H. J. Lee, Y. Lu, B. D. Freeman and I. C. Sanchez, J. Phys. Chem. B, 2006, 110, 12666–12672 CrossRef CAS.
- Y. Ichiraku, S. A. Stern and T. Nakagawa, J. Membr. Sci., 1987, 34, 5–18 CrossRef CAS.
- N. Morliere, C. Vallieres, L. Perrin and D. Roizard, J. Membr. Sci., 2006, 270, 123–131 CrossRef CAS.
- K. Nagai, A. Higuchi and T. Nakagawa, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 289–298 CrossRef CAS.
-
I. Pinnau and Z. He, Membrane Technology and Research Inc., US Pat. 6
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 316684, 2001.
316684, 2001. - T. C. Merkel, B. D. Freeman, R. J. Spontak, Z. He, I. Pinnau, P. Meakin and A. J. Hill, Science, 2002, 296, 519–522 CrossRef CAS.
- T. C. Merkel, Z. He, I. Pinnau, B. D. Freeman, P. Meakin and A. J. Hill, Macromolecules, 2003, 36, 6844–6855 CrossRef CAS.
- T.-S. Chung, L. Y. Jiang, Y. Li and S. Kulprathipanja, Prog. Polym. Sci., 2007, 32, 483–507 CrossRef CAS.
- Y. P. Yampolskii, Russ. J. Gen. Chem., 2009, 79, 657–665 CrossRef CAS.
- M. Rudel, J. Kruse, K. Raetzke, F. Faupel, Y. P. Yampolskii, V. P. Shantarovich and G. Dlubek, Macromolecules, 2008, 41, 788–795 CrossRef CAS.
- A. V. Tokarev, G. N. Bondarenko and Y. P. Yampol'skii, Vysokomol Soedin., Seriya A i Seriya B, 2007, 49, 1510–1523 Search PubMed.
- E. S. Finkelshtein, M. L. Gringolts, N. V. Ushakov, V. G. Lakhtin, S. A. Soloviev and Y. P. Yampol'skii, Polymer, 2003, 44, 2843–2851 CrossRef CAS.
- N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem.–Eur. J., 2005, 11, 2610–2620 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- N. B. McKeown, S. Makhseed and P. M. Budd, Chem. Commun., 2002, 2780–2781 RSC.
- N. B. McKeown, S. Hanif, K. Msayib, C. E. Tattershall and P. M. Budd, Chem. Commun., 2002, 2782–2783 RSC.
- A. V. Maffei, P. M. Budd and N. B. McKeown, Langmuir, 2006, 22, 4225–4229 CrossRef CAS.
- H. J. Mackintosh, P. M. Budd and N. B. McKeown, J. Mater. Chem., 2008, 18, 573–578 RSC.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456–459 CrossRef CAS.
- J. Song, N. Du, Y. Dai, G. P. Robertson, M. D. Guiver, S. Thomas and I. Pinnau, Macromolecules, 2008, 41, 7411–7417 CrossRef CAS.
- N. Du, J. Song, G. P. Robertson, I. Pinnau and M. D. Guiver, Macromol. Rapid Commun., 2008, 29, 783–788 CrossRef CAS.
- M. Heuchel, D. Fritsch, P. M. Budd, N. B. McKeown and D. Hofmann, J. Membr. Sci., 2008, 318, 84–99 CrossRef CAS.
- R. Lima de Miranda, J. Kruse, K. Raetzke, F. Faupel, D. Fritsch, V. Abetz, P. M. Budd, J. D. Selbie, N. B. McKeown and B. S. Ghanem, Phys. Status Solidi RRL, 2007, 1, 190–192 Search PubMed.
- V. P. Shantarovich, T. Suzuki, Y. P. Yampol'skii, P. Budd, V. V. Gustov, I. B. Kevdina, A. V. Pastukhov, S. S. Berdonosov and V. E. Bozhevol'nov, High Energy Chem., 2007, 41, 370–380 CrossRef CAS.
- N. Du, G. P. Robertson, J. Song, I. Pinnau and M. D. Guiver, Macromolecules, 2009, 42, 6038–6043 CrossRef CAS.
- M. Carta, K. J. Msayib, P. M. Budd and N. B. McKeown, Org. Lett., 2008, 10, 2641–2643 CrossRef CAS.
- M. Carta, K. J. Msayib and N. B. McKeown, Tetrahedron Lett., 2009, 50, 5954–5957 CrossRef CAS.
- B. S. Ghanem, N. B. McKeown, P. M. Budd and D. Fritsch, Macromolecules, 2008, 41, 1640–1646 CrossRef CAS.
- N. Du, G. P. Robertson, I. Pinnau, S. Thomas and M. D. Guiver, Macromol. Rapid Commun., 2009, 30, 584–588 CrossRef CAS.
- N. Du, G. P. Robertson, J. Song, I. Pinnau, S. Thomas and M. D. Guiver, Macromolecules, 2008, 41, 9656–9662 CrossRef CAS.
- N. Du, G. P. Robertson, I. Pinnau and M. D. Guiver, Macromolecules, 2009, 42, 6023–6030 CrossRef CAS.
- M. L. Cecopieri-Gomez, J. Palacios-Alquisira and J. M. Dominguez, J. Membr. Sci., 2007, 293, 53–65 CrossRef CAS.
- K. Tanaka, M. Okano, H. Toshino, H. Kita and K. Okamoto, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 907–914 CrossRef CAS.
- Y. Liu, C. Pan, M. Ding and J. Xu, Polym. Int., 1999, 48, 832–836 CrossRef CAS.
- W.-H. Lin, R. H. Vora and T.-S. Chung, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 2703–2713 CAS.
- M. Al-Masri, H. R. Kricheldorf and D. Fritsch, Macromolecules, 1999, 32, 7853–7858 CrossRef CAS.
- Q. Zhang, G. Chen and S. Zhang, Polymer, 2007, 48, 2250–2256 CrossRef CAS.
- H. B. Park, C. H. Jung, Y. M. Lee, A. J. Hill, S. J. Pas, S. T. Mudie, E. Van Wagner, B. D. Freeman and D. J. Cookson, Science, 2007, 318, 254–258 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |