How controlled and versatile is N-carboxy anhydride (NCA) polymerization at 0 °C? Effect of temperature on homo-, block- and graft (co)polymerization†
Gijs J. M.
Habraken
a,
Maloes
Peeters
a,
Carin H. J. T.
Dietz
a,
Cor E.
Koning
a and
Andreas
Heise
*ab
aEindhoven University of Technology, Department of Polymer Chemistry, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: a.heise@tue.nl; Fax: + 31 (0)46 4763949; Tel: + 31 (0)46 4761119
bDublin City University, School of Chemical Sciences, Glasnevin, Dublin, 9, Ireland
First published on 27th January 2010
Abstract
We have investigated the polymerization of various amino acid N-carboxyanhydrides (NCAs) at 0 °C. Detailed MALDI-ToF analysis of homopolymerizations of three amino acid NCAs clearly confirms that frequently occurring end-group termination and other side-reactions are absent at 0 °C. The polymerization is thus controlled and homo and copolypeptides with low polydispersities, around 1.1, were obtained for soluble polypeptides. MALDI-ToF contour plot analysis confirmed the randomness of the copolymers. The controlled character of the low temperature NCApolymerization was further verified by the successful block copolymer synthesis from polypeptide macroinitiators, which confirms the availability of the amino end-groups for chain extension. Moreover, graft copolymers were obtained by grafting of benzyl-L-glutamate NCA from lysine-containing copolymers. The formation of gels from the graft and corresponding block copolymers was investigated. While for the block copolymers interconnected polymer particles were observed by optical microscopy, the graft copolymers form an open cell structure.
Introduction
Polypeptides are very versatile materials that fulfil many important roles in natural systems. These range from mechanical reinforcing materials in skin or bone (e.g.collagen) to much more complex roles, for example as biocatalysts (enzymes) and in biological recognition processes like antibody/antigen interaction. Characteristic, and in fact highly important for their activity, is the defined order of the amino acid monomers of natural polypeptides and the perfect polydispersity index of 1. Due to the high potential of various polypeptides also in technical processes, biotechnological methodologies have been developed to synthesize them industrially at large scale.1 However, the development of such a process can be very tedious and requires specific expertise. Synthesis of polypeptides with a well-defined amino acid sequence and structure can be achieved on laboratory scale by a one-by-one amino acid addition on a solid support (Merrifield synthesis) but this is only possible up to limited molecular weights. If polypeptides without a specific amino acid sequence are needed, the polymerization of amino acid N-carboxyanhydrides (NCA) is a useful alternative. This technique produces high yields of polypeptides with a wide range of molecular weights. Amino acid NCA monomers are readily accessible, mostly using techniques involving phosgenation of protected amino acids or by alternative methods.2–5 While the polymerization of NCAs is generally straightforward, for example fast polymerization can be achieved by the addition of a base or nucleophilic initiator, controlling the polymerization and the molecular weight of polymers is by no means trivial.6–8 Similar to other polymerizations, control can only be achieved if the polymer end-group is the only initiating species, if its activity remains high and if termination is virtually absent. In general, initiation by a primary amine fulfils these requirements but unfortunately side reactions frequently occur involving the more basic amines, which easily abstract the proton from an NCA monomer resulting in initiation by the NCA anion monomer (activated monomer mechanism). Water contaminants can also initiate the NCA as a nucleophile, making it crucial to work under dry conditions. Another issue is the low solubility of some polypeptides, resulting in partial precipitation of polymer chains during the polymerization. The reactive polymer chain ends are then not accessible any more for propagation, whilst the well-dissolved chains continue to grow resulting in a broad polydispersity. Moreover, solvent-induced reactions have been shown to occur for certain monomers with DMF and NMP.9–11Several pathways preventing chain end termination reactions in NCApolymerizations have recently been developed.6–8 Deming introduced a nickel complex for the insertion of NCA monomers leaving the end group intact and even enabling block copolymerization.12 In another method, ammonium halides were used for creating a dormant chain end, which can reversibly transform into amine and hydrogenhalogens at higher temperatures.13,14 Another method to prevent termination is by initiation of the NCApolymerization by silazane.15 Other successful approaches rely on the alteration of the polymerization conditions in otherwise traditional primary amine-initiated NCApolymerizations. Very recently, Gibson and Cameron demonstrated that NCApolymerization by amine initiation can lead to a high level of control and even block copolymers.16 Hadjichristidis and co-workers showed that, using highly purified chemicals and clean vacuum techniques, synthesis of polypeptides of high and controlled molecular weight was possible.17 By lowering the reaction temperature of the polymerization to 0 °C Vayaboury and co-workers showed by capillary electrophoresis that the amine end group remained present at 99% of the polypeptide chains.18–20 It was suggested that predominantly the termination reaction with the used solvent (DMF) resulting in a formamide formation decreased at 0 °C. While reaction times are naturally longer at lower temperatures, this latter method is extremely promising since the reaction conditions are simple and scalable. It does not require the use of specific catalysts and or demanding reaction conditions like high vacuum technology are required. We have recently reported that this method can be applied to the synthesis of well-defined poly(γ-benzyl-L-glutamate) (PBLG) macroinitiators for controlled radical polymerizations and for the synthesis of P(BLG-co-cysteine).21,22 However, up to now its wider applicability for the synthesis of a large variety of polypeptides from different amino acid NCAs has not been shown. In this work we systematically investigate the factors influencing the level of control achievable in the low reaction temperature method for a range of NCA monomers in homo and copolymerizations. Special emphasis was placed on establishing the effect of the polymerization temperature on the polymer structure in homo and copolymerizations and thus the level of control under these reaction conditions. Moreover, the applicability of the synthesis of polymer architectures, such as block and graft copolymers was investigated.
Experimental
Materials
Benzylamine 99.5% purified by redestillation, S-tert-butylmercapto-L-cysteine, S-benzyl-L-cysteine, L-alanine, α-pinene 98%, bis(trichloromethyl) carbonate (triphosgene) 99%, piperidine 99% and trifluoroacetic acid 99% were purchased from Aldrich. L-Glutamic acid, γ-benzyl ester, L-aspartic acid, β-benzyl ester, O-benzyl-L-serine, Nε-benzyloxycarbonyl-L-lysine, Nε-fluorenylmethoxycarbonyl-L-lysine and N-trityl-L-glutamine were supplied by Bachem. DMF (extra dry), ethylacetate, n-heptane and diethylether were purchased from Biosolve. All chemicals were used without any purification unless mentioned. DMF and ethylacetate were used directly from the bottle or stored under an inert, dry atmosphere. NCAs were synthesized as described elsewhere unless otherwise noted.23Methods
For the SEC analysis using HFIP (Biosolve, AR-S from supplier or redistilled) as eluent measurements were done using a Shimadzu LC-10AD pump (flow rate 0.8 ml min−1) and a WATERS 2414, differential refractive index detector (at 35 °C). Injections were done by a Spark Holland, MIDAS injector, for which a 50 μL injection volume was used. The column was a PSS, 2* PFG-lin-XL (7 μm, 8 × 300 mm) column, used at 40 °C. Calibration has been done using poly(methyl methacrylate) standards.For the SEC analysis using DMF (Biosolve) as eluent measurements were done on a Waters Alliance system equipped with a Waters 2695 separation module, a Waters 2414 refractive index detector (40 °C), a Waters 486 UV detector, a PSS GRAM guard column followed by 2 PSS GRAM columns in series of 100 (10 mm particles) and 3000 (10 mm particles) respectively at 60 °C. DMF was used as eluent at a flow rate of 1 mL min−1. The molecular weights were calculated using polystyrene standards. Before SEC analysis is performed, the samples were filtered through a 0.2 μm PTFE filter (13 mm, PP housing, Alltech).
1H-NMR analyses were performed on a Mercury 400. 13C-NMR analyses were performed on an Inova 500. For the monomers deuterated chloroform was used. For the polymersDMSO-d6 and deuterated TFA were used. For the composition of the copolymers (Table 3) the composition was determined by comparing the integral values of γ-benzyl-L-glutamate 2.2 ppm (2H), O-benzyl-L-serine 3.7 ppm (2H), L-alanine 1.5 ppm (3H) and FMOC-L-lysine 1.3 ppm (4H). For the copolymer of trityl-L-glutamine and γ-benzyl-L-glutamate the integral value for γ-benzyl-L-glutamate at 5.0 ppm (2H) was subtracted from total integral of all the aromatic groups (7.4 ppm).
Matrix assisted laser desorption/ionitiation–time of flight–mass spectroscopy analysis was carried out on a Voyager DE-STR from Applied Biosystems (laser frequency 20 Hz, 337nm and a voltage of 25kV). The matrix material used was DCTB (40 mg ml−1). Potassium trifluoroacetic acid (KTFA) was added as cationic ionization agent (5 mg ml−1). The polymer sample was dissolved in HFIP (1 mg ml−1), to which the matrix material and the ionization agent were added (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5), and the mixture was placed on the target plate. Samples were precipitated from the reaction medium in diethylether, filtered and placed in a freezer before measuring.
:1:5), and the mixture was placed on the target plate. Samples were precipitated from the reaction medium in diethylether, filtered and placed in a freezer before measuring.
Samples for microscopy were prepared by dissolving the polymers (1.0 mg) in 0.1 g TFA and subsequently in 10 ml CHCl3. The solution was put on a glass plate or grid and dried under an argon flow. The solvent was finally removed by vacuum and the sample was sputtered for 3 min. Dark field optical microscopy was done with a Zeiss axioplan 2 using an LD epiplan 50x objective.
Synthesis
Results and discussion
Monomer synthesis
The NCA monomers were synthesized following a standard literature procedure by reacting the (protected) amino acids with triphosgene in the presence of an HCl scavenger such as α-pinene or triethylamine (Scheme 1).3,24 The synthesis of the NCA of N-trityl-L-glutamine has not been reported before. When the standard procedure was applied the NCA of 2-amino-4-cyano-(S)-butanoic acid was formed.25 Since the trityl group is removed in the presence of HCl, the free amide can convert to the nitrilegroup by reaction with phosgene. By altering the concentration of α-pinene and decreasing the reaction time to under an hour the product could be isolated as a white powder. All monomers were obtained in high purity after repeated recrystallization from an ethyl acetate n-heptane solution.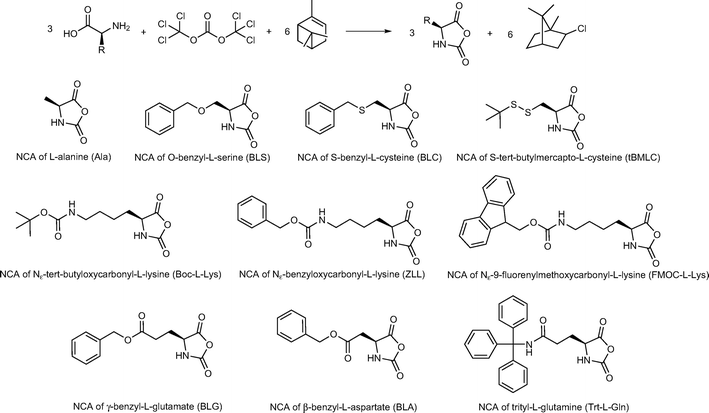 | ||
| Scheme 1 List of amino acid N-carboxy anhydrides (NCAs) applied in the polymerizations at 0 °C. | ||
Homopolymerization
The effect of temperature on the NCA ROP was determined for the polymerization of three NCAs, i.e.γ-benzyl-L-glutamate (BLG), β-benzyl-L-aspartate (BLA) and O-benzyl-L-serine (BLS). All polymerizations were initiated with benzylamine (monomer to initiator ratio (M/I) = 40) in DMF and carried out at four different temperatures (0, 10, 20 and 60 °C) for four days under an inert atmosphere. After precipitation, the polymers were analyzed by size exclusion chromatography (SEC) and MALDI-ToF-MS. SEC does not reveal a conclusive effect of the reaction temperature on the obtained molecular weights. While within reasonable variations the molecular weights are comparable for a temperature series (Table 1) direct comparison between expected and experimental molecular weight cannot be made since the monomer conversions of the individual polymerizations are unknown. Noteworthy is that the polydispersity index (PDI) of the PBLG sample obtained at 60 °C (1.27) is higher than the PDI obtained at lower temperatures (1.06–1.13) and that all PBLS samples have a bimodal distribution.All final polymer samples were further analyzed by MALDI-ToF-MS to determine the chemical composition (for all spectra see the supporting information). For PBLG it is known that pyroglutamategroups can be formed by intramolecular cyclization of the amino end-group with the adjacent benzyl ester.26 The effect of the reaction temperature on this reaction is clearly visible from the PBLGspectra shown in Fig. 1. While at 0 °C only PBLGmacromolecules with intact amino end-groups are detected (structure A, Fig. 2) the relative amount of pyroglutamate (structure B, Fig. 2) successively increases with increasing temperature. At 20 °C already a substantial fraction of the chains are terminated by pyroglutamate and at 60 °C all end-groups are pyroglutamates. The kinetics of this chain end termination cannot be concluded from these measurements, but it is reasonable to assume that it happens throughout the polymerization and leads to an increasing number of dead chain-ends as the polymerization proceeds. This would explain the higher polydispersity observed for the PBLG obtained at 60 °C as compared to the polymers obtained at lower temperature.
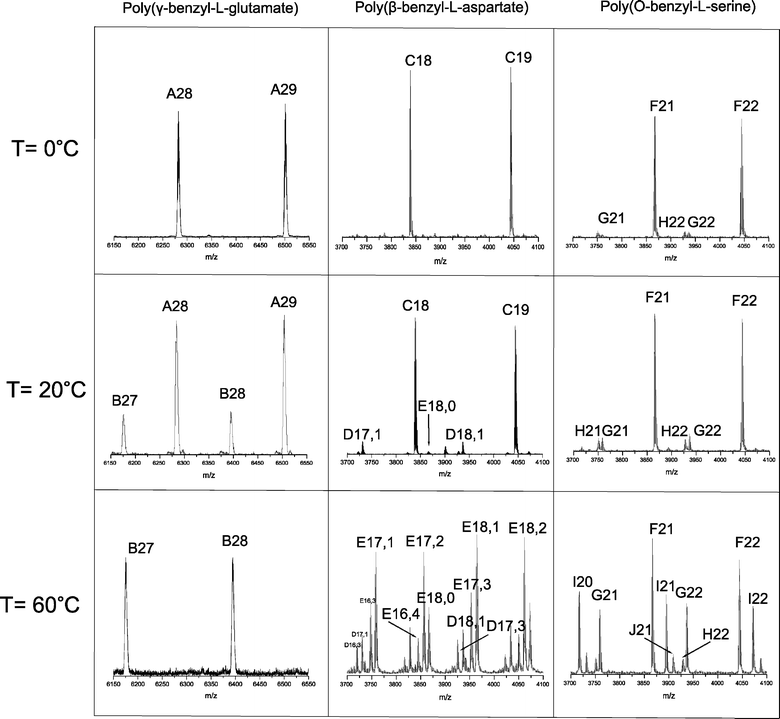 | ||
| Fig. 1 MALDI-ToF-MS results of PBLG, PBLA and PBLS obtained at different temperatures. Letters in peak assignments refer to structures shown in Fig. 2; numbers denote the degree of polymerization. For the poly(β-benzyl-L-aspartate) the first number refers to the β-benzyl-L-aspartate units and the second number to the anhydrides units in the formed copolymers. All samples were measured with potassium trifluoroacetic acid (KTFA). | ||
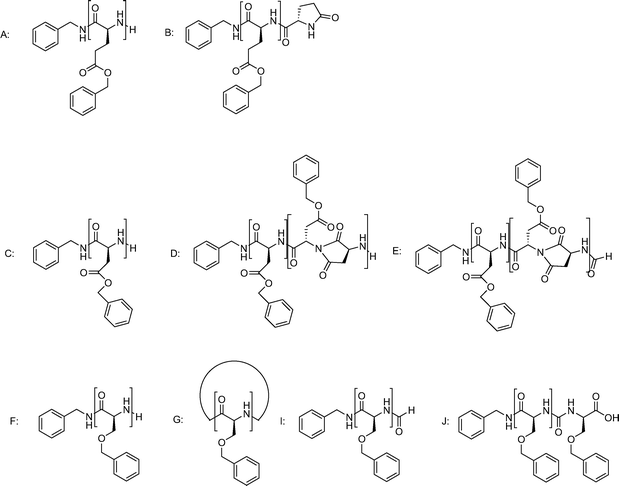 | ||
| Fig. 2 Structures identified in the NCApolymerization of BLG, BLA and BLS. The letters refer to the MALDI-ToF peak assignment in Fig. 1. | ||
Also for the PBLA the MALDI-ToFspectrum for the polymer obtained at 0 °C confirms the presence of only one polymer species, namely PBLA with amino end-groups (structure C, Fig. 2). At higher temperatures the spectra become more complex with a series of peaks suggesting the formation of copolymers. These side reactions are the result of an intramolecular amidation leading to the formation of succinimide units in the main chain (structure D, Fig. 2). For the reaction at 60 °C PBLA with at least five succinimide units per chain could be identified. An increasing amount of formamidegroups was also found at the chain ends (structure E, Fig. 2) in addition to the succinimide formation. This chain end termination is the result of the reaction of the amine end-group with the solventDMF, yielding formamide terminated polymer chains and dimethylamine.9 As an end-capping reaction, the formamide formation will terminate polymergrowth. However, compared to the chain-end termination of the PBLG, the effect on the molecular weight and polydispersity seems to be minor. The presence of the succinimidegroups could be more problematic if the polymer comes into contact with water, which could result in a ring opening reaction producing either a normal α amide or a β-peptide bond.27
For the PBLS some side products can be determined even for the polymerization at 0 °C. Besides the benzylamine-initiated and unterminated chains (structure F, Fig. 2) also cyclic structures (structure G, Fig. 2) and a so far unidentified species (structure H, Fig. 2) were found.10 The full MALDI-ToF-MS spectrum (ESI†) shows that for the lower degrees of polymerization a relatively larger amount of cyclic structures (structure G, Fig. 2) is present, which implies that there is a molecular weight dependency. At higher temperature in particular an increase of the relative amounts of larger cyclic structures is observed. Moreover, at 60 °C other side reactions occur, for example formamide formation by reaction with DMF (structure I, Fig. 2). Structure J (Fig. 2) was identified as a product of a polymer chain, which had reacted with isocyanatocarboxylic acid. The latter is most likely formed at the higher temperature from the deprotonated NCA resulting in the so-called hydantoic acid end group.11
In conclusion, for all investigated samples a significant effect of the reaction temperature on the polymer structure was observed. With the exception of PBLG, which shows an increase in PDI at higher reaction temperatures, these effects are not evident from SEC. Striking is the difference between the side reactions of the three investigated polymers. For the PBLG only end-group termination occurs, while for PBLA succinimide formation and end-group termination by reaction with DMF was detected. The latter was also observed for PBLS alongside with cyclization reactions. Without exception, the best results were obtained for the polymerizations performed at 0 °C resulting in high structural control with retention of the active amino end-groups. It has to be noted that all samples were treated identically. The fact that no side-reactions were detected at 0 °C thus excludes any post-polymerization or MALDI-ToF effect on the results.
The feasibility of the NCApolymerization at 0 °C in DMF was then investigated for a series of other monomers. With a few exceptions these polymerizations resulted in polymers with a low PDI and a molecular weight as targeted between 4,000 and 9,000 g mol−1 (Table 2). It has to be noted that due to the calibration of the SEC with PMMA a quantitative comparison with the theoretical molecular weight is difficult. For the L-alanine and the S-tert-butylmercapto-L-cysteineNCA a high PDI (> 2.0) was obtained, suggesting a limited control over the reaction. This is due to the observed precipitation or aggregation of polymers during the polymerization. MALDI-ToFspectra of poly(S-tert-butylmercapto-L-cysteine) also confirmed side reactions such as cyclization and chain end termination.
| Entry | Polymer | M/I | Mntheo/g mol−1c | Mn/g mol−1 | PDI |
|---|---|---|---|---|---|
| a Measured with DMF-SEC, calibrated with polystyrene standards. b Results published elsewhere.23 c Calculated from the M/I ratio. | |||||
| 1 | Poly(γ-benzyl-L-glutamate) | 41 | 9,100 | 9,300 | 1.1 |
| 2 | Poly(Nε-t-Boc-L-lysine) | 40 | 9,200 | 6,500 | 1.2 |
| 3 | Poly(Nε-Z-L-lysine) with LiBr | 14 | 3,800 | 8,100 | 1.1 |
| 4 | Poly(Nε-Fmoc-L-lysine) a | 20 | 7,100 | 5,100 | 1.3 |
| 5 | Poly(O-benzyl-L-serine) | 12 | 2,200 | 6,200 | 1.1 |
| 6 | Poly(S-benzyl-L-cysteine) b | 15 | 3,000 | 4,700 | 1.1 |
| 7 | Poly(S-t-butylmercapto-L-cysteine) | 18 | 3,500 | 3,900 | 2.0 |
| 8 | Poly(N-Trityl-L-Glutamine) | 35 | 13,100 | 6,300 | 1.1 |
| 9 | Poly(β-Benzyl-L-aspartate) | 40 | 8,300 | 7,300 | 1.1 |
| 10 | Poly(L-alanine) | 40 | 2,900 | 4,400 | 2.2 |
Copolymerization
In order to investigate the applicability of the low temperature synthesis for copolymerizations, BLG was copolymerized with several NCAs. Both comonomers were dissolved in DMF at 0 °C, followed by the addition of a solution of DMF containing benzylamine. After a reaction time of four days the polymer solutions were precipitated in diethylether and the obtained polymer investigated by SEC and MALDI-ToF. Initially the effect of the monomer feed composition on the molecular weights and final composition of the copolymer was examined for BLS and BLG. Three experiments were carried out with comonomer ratio of 1:3, 1:1 and 3:1 (Table 3, entries 1–3). A precipitation was observed for the polymerization with higher BLS quantities (entries 2 and 3). The obtained molecular weights were comparable for all three reactions (8,300 – 11,000 g mol−1), but a bimodal distribution was obtained for the partially precipitated samples. This also resulted in a higher PDI of >1.2 as compared to the 1.1 of the homogeneous reaction (entry 1). All other reactions were homogeneous and produced polymers with PDIs of around 1.2. For the N-trityl-L-glutamine and γ-benzyl-L-glutamate copolypeptide a lower than expected molecular weight was found, which was also observed for the N-trityl-L-glutaminehomopolymer (Table 2). This might possibly be due to the bulkiness of the trityl-sidegroup.
| Entry | M1 | M2 | Feed ratio | Mn/g mol−1 | PDI | Composition (1H-NMR) |
|---|---|---|---|---|---|---|
| a Measured by DMF-SEC with PS standards. | ||||||
| 1 | BLS | BLG | 10:30 |
10,300 | 1.1 | 1.0:3.6 |
| 2 | BLS | BLG | 20:20 |
11,000 | 1.2 | 1.0:1.0 |
| 3 | BLS | BLG | 30:10 |
8,300 | 1.3 | 2.0:1.0 |
| 4 | Ala | BLG | 20:20 |
8,600 | 1.1 | 1.0:1.0 |
| 5 | FMOC-L-Lysa | BLG | 10:41 |
8,600 | 1.2 | 1.0:3.5 |
| 6 | FMOC-L-Lysa | BLG | 9:37 |
7,800 | 1.1 | 1.0:2.6 |
| 7 | FMOC-L-Lys | BLG | 20:59 |
12,700 | 1.2 | 1.0:2.3 |
| 8 | Gln(Trt) | BLG | 19:18 |
3,500 | 1.2 | 1.0:1.2 |
The MALDI-ToF analysis of the copolymers was in agreement with the results obtained for the homopolymers. For copolymerizations performed at 0 °C no side reaction products were found by MALDI-ToF-MS with the exception of the BLS copolymerization. As in the homopolymerization an increasing relative ratio of cycles was seen with increasing amount of BLS in the monomer feed.
A first indication of the monomer composition in the copolymers was obtained from 1H-NMR (Table 3). Generally good agreement was found between the monomer feed ratios and the compositions determined by 1H-NMR. However, this method provides only information on the overall composition of the copolymer but not on the monomer distribution along the polymer chain. While NCAcopolymerizations have been reported before, copolymerization parameters of these reactions are unknown. The reason is that these are very difficult to determine as no method is available so far that allows to follow the individual monomer conversion. We were, however, able to apply a software based MALDI-ToF deconvolution method developed in our group, which allows converting the spectra into composition contour plots.28–30 The shape of the contour plot allows drawing conclusions concerning the molecular distribution of the comonomers in the chain, i.e. whether it is a random or a block structure. Block copolymers can be identified as circular or elliptical (if the PDI of one bock is higher) shapes, where the ellipsoid has axes parallel to the X or Y-axes. For random copolymers the circular shape can be found for a controlled system with a higher reactivity ratio for one of the monomers or for a Bernoulli random copolymer (r1 = r2 = 1, 50% composition). Otherwise an elliptical shape is found for copolymers where a directional coefficient goes through zero.30 For example, for the contour plot of copolymer 1 (Table 3) a single distribution was found to exhibit a maximum for 25 BLG and 7 BLS units (Fig. 3a). This is characteristic for a random copolymer and in agreement with the monomer feed ratio. From the directional coefficient of the contour plot of copolymer 2 (Fig. 3b) a random copolymerization with a monomer composition of 1:1 can be concluded, again in agreement with the feed ratio. Similarly, the contour plot of copolymer 3 confirms the random structure with the expected comonomer composition (Fig. 3c). It has to be noted, that due to the mass discrimination in the MALDI-ToF-MS spectra the bimodal character of the distribution is increased and the composition of the maxima cannot directly be determined from the contour plots.30
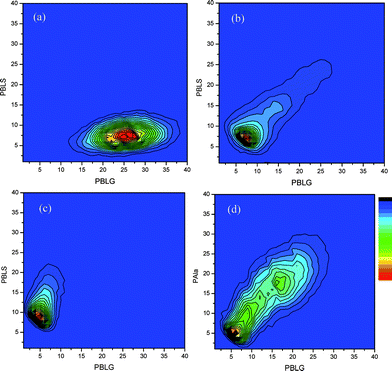 | ||
| Fig. 3 MALDI-ToF-MS contour plots of P(BLS-co-BLG) for monomer feed ratios 10:30 (a), 20:20 (b) and 30:10 (c) (Table 3, entries 1–3) and P(Ala-co-BLG) (d) ((Table 3, entry 4). | ||
A similar contour plot was calculated from a MALDI-ToFspectrum of P(BLG-co-Ala) (entry 4, Table 3) with a targeted composition of 20:20 (Fig. 3d). The SEC of this copolymer showed a Gaussian distribution with a low polydispersity and no side-reactions were detected in the MALDI-ToF-MS. As for the P(BLG-co-BLS) (entry 2, Table 3) the contour plot confirms that the composition is in the range of the weighed-in 50:50 ratio with a random monomer distribution. The results provide evidence that the copolymerization at 0 °C results in random copolymers with comonomer ratios close to the monomer feed ratios.
Block copolymerization
Block copolymer synthesis by sequential monomer addition (macroinitiation) is ideally suited to investigating the degree of control of a polymerization. A successful macroinitiation is dependent on the presence of active chain ends on the polymer after the first polymerization and on an efficient initiation step. Any deactivation of chain ends or inefficient macroinitiation will inevitably result in unreacted macroinitiator and thus in homopolymer residues. Our results so far confirmed that the NCApolymerization at 0 °C fulfils the first requirement, i.e.polypeptides with active amino end-groups are obtained. In the following experiments we investigated whether this methodology can be extended to the synthesis of polymer architectures such as block and graft copolymer. This is of specific relevance since polypeptideblock copolymers have shown interesting self-assembly behavior and were applied in responsive materials.31–35We first attempted the synthesis of P(BLS-b-BLG) from a well-defined isolated PBLS macroinitiator of 6,100 g mol−1 (Table 4, entry 1). The synthesis of the blocks was performed by adding the NCA of γ-benzyl-L-glutamate to the solution of the first block for 4 days.17,36 As can be seen from the SEC plot in Fig. 4a, a block copolymer with a broad distribution (PDI = 1.89) and a significant amount of unreacted macroinitiator was obtained. PBLS is a strong β-sheet forming polymer and we hypothesized that the reaction solventDMF did not disrupt the hydrogen bonds so as to make all PBLS equally available for macroinitiation. Since PBLG is much better soluble in DMF, inversing the order of monomer addition was expected to improve the results. Indeed when BLG was polymerized first, followed by the addition of the NCA of BLS without isolation and purification of the first block, a complete shift from the macroinitiator to the block copolymer was observed in SEC (Fig. 4b). The molecular weight increased from 6,400 g mol−1 for the PBLG macroinitiator to 9,700 g mol−1 for the block copolymer with a PDI of 1.06 (Table 4, entry 4). As a control experiment macroinitiations from PBLG obtained at 20 and 60 °C were carried out. While in the latter case no increase of molecular weight was observed, macroinitiation from the PBLG obtained at 20 °C produced some block copolymer with significant amounts of PBLG left (ESI†). This is in agreement with MALDI-ToF results indicating increasing loss of end-groups with increasing polymerization temperature. Moreover, by changing the monomer ratios PBLG-b-PBLS block copolymers with various block length ratios and low polydispersities were successfully synthesized (Table 4, entries 2, 3 and 5) even when a gelation effect was seen upon the polymerization for approximately 10 wt% monomer in DMF for most entries. This seems to be contradictive with the low PDI, but the gelation occurs only after 2 to 3 days when a considerable amount of the second block is already formed. With a uniform gelation all polymerizations are quenched, due to a lack of chain mobility.
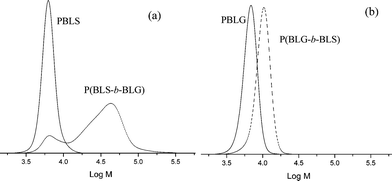 | ||
| Fig. 4 SEC (HFIP) results of P(BLS-b-BLG) synthesis by sequential monomer addition. (a) Addition of BLGNCA to a PBLS macroinitiator (entry 1, Table 4) and (b) addition of BLS NCA to a PBLG macroinitiator (entry 4, Table 4). | ||
| Entry | Block 1 | Block 2 | Block 1 | Block 2 | ||
|---|---|---|---|---|---|---|
| Mn/g mol−1 | PDI | Mn/g mol−1 | PDI | |||
| a Precipitated macroinitiator. | ||||||
| 1 | PBLS12a | PBLG24 | 6,100 | 1.1 | 19,800 | 1.9 |
| 2 | PBLG39 | PBLS15 | 8,900 | 1.1 | 12,400 | 1.1 |
| 3 | PBLG42 | PBLS33 | 9,000 | 1.1 | 13,900 | 1.1 |
| 4 | PBLG20 | PBLS20 | 6,400 | 1.1 | 9,700 | 1.1 |
| 5 | PBLG40a | PBLS40 | 10,600 | 1.1 | 15,800 | 1.1 |
| 6 | PBLG44 | PBLC16 | 8,800 | 1.1 | 12,800 | 1.1 |
| 7 | PBLG40 | PtBMLC15 | 8,100 | 1.1 | 11,700 | 1.2 |
| 8 | PBLG41 | PtBMLC41 | 16,000 | 1.1 | 21,300 | 1.4 |
| 9 | PBLG20 | PAla20 | 6,300 | 1.1 | 7,500 | 1.1 |
The MALDI-ToF contour plot of the block copolymers clearly differ from the corresponding contour plots of the random copolymers in that there is no directional coefficient (Fig. 5). The circular distribution with a pronounced maximum, as seen for the example of P(BLG-b-Ala) in Fig. 5b, is typical for block copolymers. For this block copolymer the block structure was also evident from 13C-NMR.37,38 The contour plot of P(BLS-b-BLG) shows two different maxima at the monomer composition of 15/15 and 17/17, respectively. This might indicate that there was still a small amount the first monomer present at the time of the addition of the second monomer, which might give rise to some copolymerization in the second block. This could be due to the gelation in the reaction mixture, which was seen at the end of the first polymerization.
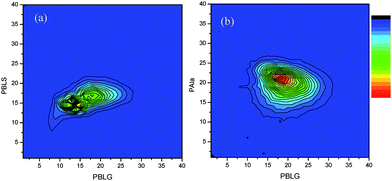 | ||
| Fig. 5 (a) Contourplot of MALDI-ToF-MS spectrum of P(BLG-b-BLS) (entry 4, Table 4) (b) Contourplot of MALDI-ToF-MS spectrum of P(BLG-b-Ala) (entry 9, Table 4). | ||
The block copolymer synthesis by addition of different cysteine NCAs to the PBLG macroinitiator was successful when a low degree of polymerization was targeted for the cysteine block. For higher degrees of polymerization the polydispersities increased for the S-tert-butylmercapto-L-cysteine and a bimodal distribution was visible.
Graft polymerization
For the manufacturing of graft copolymers the copolymers of γ-benzyl-L-glutamate and N-FMOC-L-lysine (Table 3, entry 5) were selectively deprotected, resulting in a backbone of PBLG and L-lysine with functional amines present as pendant groups for further polymerization (Scheme 2).24,39,40 Approximately 200 mg of deprotected copolymer was dissolved in DMF and different amounts of BLG and BLS NCA were added. Fig. 6 shows the clear increase in molecular weight, corresponding to the ratio of NCA to the P(BLG-co-Lys) (Table 5). The PDI of the graft copolymers increases to 1.6–1.9, which suggests a somewhat higher PDI index of the grafted blocks. This increase in the PDI can be caused by the different number of initiating amine groups in the chain due to the random character of the polymer. Moreover, SEC traces reveal a peak at lower molecular weight which is believed to be caused by a secondary initiation mechanism. Indeed MALDI-ToF-MS confirms that pure PBLG is present in these samples. Whether this is caused by impurities in the reaction mixture or by initiation by the activated monomer mechanism from the sterically hindered amines present along the grafted main chain cannot be concluded from the obtained data.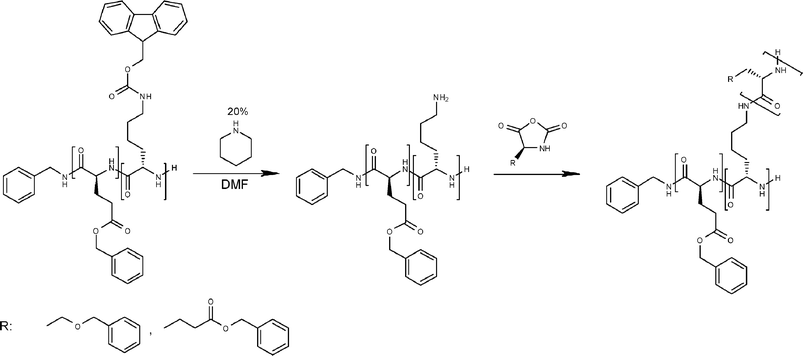 | ||
| Scheme 2 Polymerization and deprotection reactions for grafted NCA-prepared polypeptide. | ||
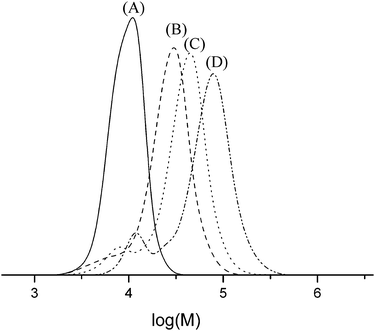 | ||
| Fig. 6 SEC traces of (A) P(BLG-co-FMOCLLys), (B) P(BLG40-co-(Lys10-g-PBLG10), (C) P(BLG40-co-(Lys10-g-PBLG20) and (D) P(BLG40-co-(Lys10-g-PBLG40) measured by DMF-SEC calibrated with polystyrene standards. Subscript numbers denote the targeted degree of polymerization. | ||
Polymer gels
Block and graft copolymers of PBLG and PBLS form gels at concentrations of around 10 wt% in DMF. As reported earlier for similar block copolymers this is due to an α-helix and β-sheet formation in the solvent.33 To compare the self-assembly structures of the block and grafted copolymers, a solution of < 0.1 mg ml−1 of the polymers in chloroform containing 1 vol% TFA was placed on a glass slide. TFA was added to disrupt the hydrogen bonds between the polymer chains of the otherwise insoluble polymer. Dark field optical microscopy did show a difference between the obtained structures (Fig. 7). For the block copolymers (Table 4, entry 4 and 5) two types of structures were found, namely a monolayer and a dense three-dimensional structure. During the evaporation of the solvent a gel formed in the concentrated regime, caused by the hydrogen bonding between the O-benzyl-L-serine blocks. For the corresponding graft copolymer of γ-benzyl-L-glutamate and L-lysine (Table 5, entry 4), on the other hand, completely different structures were obtained. The polymers resemble a three-dimensional open structure of interconnected fibers on the glass slide (Fig. 7). | ||
| Fig. 7 Optical microscopy pictures, from left to right: P(BLG-b-BLS) entry 4 Table 4, P(BLG-b-BLS), entry 5 Table 4, P(BLG-co-(Lys-g-PBLS)) entry 4, Table 5. | ||
Conclusions
We have investigated the polymerization of various amino acid NCAs at 0 °C. Detailed MALDI-ToF analysis clearly confirms the positive effect of the lower reaction temperature on the frequency of end-group termination and side-reactions. In all investigated cases the homo and copolymerizations at 0 °C were controlled and resulted in well-defined polypeptides with low polydispersities, with the exception of polymers with low solubility in the reaction medium. MALDI-ToF contour plot analysis further confirmed the randomness of the copolymerizations. The controlled character of the low temperature NCApolymerization was further verified by the successful block copolymer synthesis from polypeptide macroinitiators, which confirms the availability of amino end-groups for chain extension. Moreover, graft copolymers were obtained by NCA grafting from well-defined lysine containing copolypeptides. These polymer form gels in DMF with a different morphology as compared to the block copolymers.The presented results confirm that NCApolymerization at 0 °C is a simple and feasible method to obtain a variety of well-defined homo- and copolypeptides as well as more complex polypeptide architectures.
Acknowledgements
This work forms part of the research program of the Dutch Polymer Institute (project #610). The authors want to thank Martin Fijten for the size exclusion measurements, Friso v.d. Woerdt for some of the MALDI-ToF-MS experiments. A.H. thanks the Science Foundation Ireland (SFI) for financial support.References
- J. van Hest and D. A. Tirell, Chem. Commun., 2001, 1897–1904 RSC.
- N. M. B. Smeets, P. L. J. van der Weide, J. Meuldijk, J. A. J. M. Vekemans and L. A. Hulshof, Org. Process Res. Dev., 2005, 9, 757–763 Search PubMed.
- F. Corneille, J.-L. Copier, J.-P. Senet and Y. Robin, EP 1201659, 2002.
- W. Vayaboury, O. Giani, H. Collet, A. Commeyras and F. Schué, Amino Acids, 2004, 27, 161–167 CrossRef CAS.
- Y. Fujita, K. Koga, H.-K. Kim, X.-S. Wang, A. Sudo, H. Nishida and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5365–5370 CrossRef CAS.
- T. J. Deming, Adv. Polym. Sci., 2006, 202, 1–18 CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitskalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS.
- H. R. Kricheldorf, C. von Lossow and G. Schwarz, Macromolecules, 2005, 38, 5513–5518 CrossRef CAS.
- H. R. Kricheldorf, C. von Lossow and G. Schwarz, Macromol. Chem. Phys., 2005, 206, 282–290 CrossRef CAS.
- H. R. Kricheldorf, C. von Lossow and G. Schwarz, Macromol. Chem. Phys., 2004, 205, 918–924 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS.
- I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944–2945 RSC.
- J.-F. Lutz, D. Schuett and S. Kubowicz, Macromol. Rapid Commun., 2005, 26, 23–28 CrossRef CAS.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115 CrossRef CAS.
- M. I. Gibson and N. R. J. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2882–2891 CrossRef CAS.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS.
- W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schué, Macromol. Rapid Commun., 2004, 25, 1221–1224 CrossRef CAS.
- W. Vayaboury, O. Giani, H. Cottet, S. Bonaric and F. Schué, Macromol. Chem. Phys., 2008, 209, 1628–1637 CrossRef CAS.
- C. Scholz, V. Vayaboury, WO 2009058291 2009.
- G. J. M. Habraken, J. P. A Heuts, C. E. Koning and A. Heise, Chem. Commun., 2009, 3612–3614 RSC.
- J. R. I. Knoop, G. J. M. Habraken, N. Gogibus, S. Steig, H. Menzel, C. E. Koning and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3068–3077 CrossRef CAS.
- G. J. M. Habraken, C. E. Koning and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6883–6893 CrossRef CAS.
- J. Rodriguez-Hernandez and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1167–1187 CrossRef.
- M. Wilchek, S. Ariely and A. Patchornik, J. Org. Chem., 1968, 33, 1258–1259 CrossRef CAS.
- J. Johnes, in Amino Acid and Peptide Synthesis, Oxford University Press, Oxford, 1992 Search PubMed.
- V. Saudek, H. Pivcová and J. Drobník, Biopolymers, 1981, 20, 1615–1623 CrossRef CAS.
- R. X. E. Willemse, B. B. P. Staal, E. H. D. Donkers and A. M. Herk, Macromolecules, 2004, 37, 5717–5723 CrossRef CAS.
- R. X. E. Willemse and A. M. Herk, J. Am. Chem. Soc., 2006, 128, 4471–4480 CrossRef CAS.
- S. Huijser, Ph.D. Thesis, Eindhoven University of Technology, 2009.
- V. Breedveld, A. P. Povak, J. Sato, T. J. Deming and D. J. Pine, Macromolecules, 2004, 37, 3943–3953 CrossRef CAS.
- E. P. Holowka, D. J. Pochan and T. J. Deming, J. Am. Chem. Soc., 2005, 127, 12423–12428 CrossRef CAS.
- M. I. Gibson and N. Cameron, Angew. Chem., Int. Ed., 2008, 47, 5160–5162 CrossRef CAS.
- J. Rodriguez-Hernandez and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef CAS.
- J. Sun, X. Chen, C. Deng, H. Yu, Z. Xie and X. Jing, Langmuir, 2007, 23, 8308–8315 CrossRef CAS.
- S. Hanski, N. Houbenov, J. Ruokolainen, D. Chondronicola, H. Iatrou, N. Hadjichristidis and O. Ikkala, Biomacromolecules, 2006, 7, 3379–3384 CrossRef CAS.
- H. R. Kricheldorf and G. Schillig, Makromol. Chem., 1978, 179, 1175–1191 CrossRef CAS.
- H. R. Kricheldorf, Makromol. Chem., 1979, 180, 147–159 CrossRef CAS.
- J. Rodriguez-Hernandez, M. Gatti and H.-A. Klok, Biomacromolecules, 2003, 4, 249–258 CrossRef CAS.
- G. Mezö, J. Reményi, J. Kajtár, K. Barna, D. Gaál and F. Hudecz, J. Controlled Release, 2000, 63, 81–95 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: MALDI-ToF-MS spectra of homopolypeptides, of the contour plots (measured and simulated) and of the grafted copolypeptides. 13C-NMR of copolypeptides. SEC plots of block copolymerization from PBLG obtained at 20 and 60 °C. See DOI: 10.1039/b9py00337a |
| This journal is © The Royal Society of Chemistry 2010 |