Bulk charge transport in liquid-crystalline polymer semiconductors based on poly(2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene)
M.
Baklar
a,
S.
Barard
b,
D.
Sparrowe
c,
R. M.
Wilson
b,
I.
McCulloch
de,
M.
Heeney
de,
T.
Kreouzis
b and
N.
Stingelin
*ae
aDepartment of Materials, Imperial College London, London, SW7 2AZ, UK. E-mail: natalie.stingelin@imperial.ac.uk
bCentre of Material Research, Queen Mary University of London, London, E1 4NS, UK
cMerck Chemicals, Chilworth Science Park, Southampton, SO16 7QD, UK
dDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK
eCentre for Plastic Electronics, Imperial College London, London, SW7 2AZ, UK
First published on 8th July 2010
Abstract
The class of liquid-crystalline semiconducting polymers based on poly(2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene) recently has attracted significant interest in the field of organic electronics, predominantly due to their promising performance in field-effect transistor (FET) structures with device mobilities reaching—if not exceeding—those of amorphous silicon architectures. Less is known, however, about the bulk charge-transport properties of these interesting macromolecules. We therefore conducted time-of-flight (TOF) photoconductivity measurements on one particular material of this class of organic semiconductors—i.e. poly(2,5-bis(3-dodecylthiophen-2-yl)thieno[3,2-b]thiophene), pBTTT-C12—and attempted to correlate the electronic bulk properties of this polymer with its microstructural development with temperature. The effect of annealing was also investigated.
Introduction
Polymeric semiconductors have been extensively studied due to their promising electronic and optical properties.1,2 Liquid crystalline polymers are thereby a particularly interesting subclass because of the ability to influence their macro- and microscopic order by processing within the mesophase.1,3–5 Thus, it is not surprising that the liquid-crystalline poly(2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene)s, pBTTTs (Fig. 1a, inset), have attracted widespread interest.6,7 In fact, charge-carrier mobilities exceeding 1 cm2 V−1 s−1 have been demonstrated for this class of polymers in field-effect transistor (FET) devices,8,9 and blends of pBTTTs with PCBM have shown promising photovoltaic performance.10,11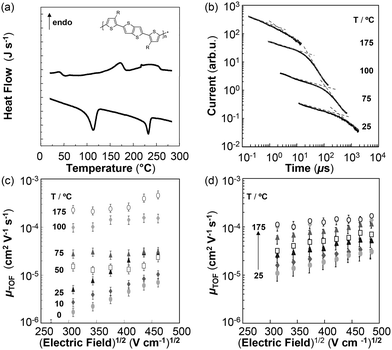 | ||
| Fig. 1 (a) Differential scanning calorimetry (DSC) first heating and cooling thermograms of as-cast pBTTT-C12 films. The inset shows the chemical structure of the pBTTT-C12 derivative utilized in this study. (b) Hole photocurrent transients across as-cast, 3.9 ± 0.7 µm-thick films of pBTTT-C12 at 46 V bias, measured at the temperatures indicated (i.e., parametric in temperature). The photocurrents are shifted vertically for sake of clarity. (c and d) Poole–Frenkel plots of hole mobility in as-cast and annealed (7 h/175 °C) pBTTT-C12 structures (film thickness of 3.9 µm and 4.7 ± 0.4 µm, respectively), which were subsequently heated from 25 °C to 175 °C (for (c) in temperature steps as indicated and in (d) in steps of 25 °C). Note that the negligible difference between mobility data obtained for annealed pBTTT-C12, measured at 100 °C and 175 °C (light grey diamond symbols and white circles, respectively), is reminiscent of the temperature independent mobilities displayed by liquid crystalline systems within a given phase (see ref. 21) and is consistent with the liquid-crystalline-like behaviour expected at elevated temperatures. | ||
Here, we focus on the poly(2,5-bis(3-dodecylthiophen-2-yl)thieno[3,2-b]thiophene), pBTTT-C12, derivative. The thermal behaviour of as-cast films of this semiconductor, as determined by differential scanning calorimetry (DSC), is shown in Fig. 1a. We selected to measure as-cast films in order to be able to obtain information on the microstructure development of such architectures during heating. The first endotherm around 50 °C is only observed on the first heating cycle, and as previously reported by DeLongchamp et al., we ascribe this to a metastable phase.12 Upon further heating a second endotherm occurs around 175 °C (peak temperature, onset is observed around 150 °C). Previous studies have demonstrated that this transition can be attributed to the melting of the side chains when entering a smectic-like phase,12 and it has been shown that brief annealing within this smectic-like phase and subsequent cooling to room temperature give rise to well defined domains based on pBTTT-C12 “lamellar stacks” that exhibit high levels of molecular order, as evidenced by specular and grazing incidence X-ray diffraction, and that result in excellent field-effect mobilites.14,15 [The transition at 230 °C has been ascribed to backbone melting.13]
It should be noted, though, that these previous studies have concentrated upon pBTTT films of a thickness in the order of 20–50 nm, which are representative of thin-film transistor devices, where performance is predominantly dictated by the molecular arrangement at the very interface to the gate dielectric. In contrast, in the present study, we were interested in the charge-transport properties of pBTTT bulk structures, i.e. charge transport out-of-plan of such film architectures, perpendicular to the lamellar stacks. Our focus thereby was on establishing relevant structure–property interrelationships, especially with regard to temperature dependence, to gain information on how the molecular arrangement in the various pBTTT-C12 solid-state phases affects bulk charge transport. In addition, we investigated how the electronic properties of this interesting polymer can be manipulated through annealing in the mesophase, similar to the heat-treatment procedures used in the fabrication of pBTTT FET devices.14,15
In a first set of experiments, we performed time-of-flight (TOF) photoconduction measurements on as-cast pBTTT-C12 films of a few micrometre thickness (sandwiched between two electrodes to form a diode structure), with photo-transients being recorded at various temperatures (Fig. 1b). Despite the relatively high dispersion evident in the photocurrents, which is a feature often observed in TOF data of polymer semiconductors and could be exacerbated in the present case due to some thickness non-uniformity of our pBTTT-C12 architectures, the inflection point arrival times (indicated with arrows in Fig. 1b) were still relatively well distinguishable for all transients. As a result, the corresponding charge-carrier mobilities µTOF could be deduced (see Materials and methods section), which allowed us to generate mobility parametrics with respect to electric field and temperature (Fig. 1c). From these, it is evident that above 50 °C the field-dependence of µTOF drastically decreased—which can be attributed to increased molecular order (see below), consistent with the less dispersive charge transport that is also observed at these temperatures. More interestingly, and in agreement with previous reports on liquid-crystalline polymer semiconductor systems,16 at higher temperatures (150 to 175 °C), in the regime where the transition to the mesophase is observed in thermal analysis, also the charge-carrier mobility was considerably affected, with µTOF reaching up to 3 × 10−4 cm2 V−1 s−1, i.e. a value more than one order of magnitude higher than that at room temperature. [Note: heating to temperatures above 175 °C resulted in electrical shorts.]
These observations on the electronic properties pBTTT-C12 can be directly correlated with the microstructural development of this polymer with temperature, as derived from wide-angle X-ray scattering (WAXS) and UV-vis spectroscopy (Fig. 2). For instance, as-cast pBTTT-C12 structures were found to be of very low molecular order giving rise to only very weak X-ray reflections (Fig. 2a) in agreement with the relatively low charge-carrier mobilities deduced from TOF. However, WAXS data recorded during heating as well as cooling cycles (Fig. 2a and b, respectively) reveal that above 50 °C transition, the wide-angle X-ray reflections at 2θ ≈ 8° intensified (Fig. 2a). When further heating to temperatures above 140 °C—the pBTTT-C12's mesophase—more intense, well-defined diffraction peaks developed consistent with the distinctly faster bulk charge transport observed in TOF, yet they were observed at slightly lower diffraction angles compared to the as-cast films at room temperature (Fig. 2a; see also ref. 12). Upon cooling, this solid–solid phase transition was found to be reversible (in accord with our DSC data). The resulting [room temperature] pBTTT-C12 structures were significantly more crystalline in comparison with as-cast films as deduced from the stronger diffraction intensities and narrower half-width at half-maxima that are observed in such heat-treated architectures, as well as the appearance of higher order diffractions (Fig. 2b). In UV-vis spectroscopy, we in addition find more distinct absorption features developing after heating pBTTT-C12 to 175 °C and subsequent cooling, indicating stronger molecular interactions in such annealed pBTTT-C12 compared to the as-cast polymer (Fig. 2c and d).17
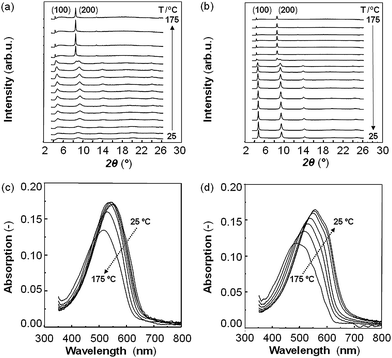 | ||
| Fig. 2 Wide-angle X-ray scattering (WAXS) diffractograms (a, b) and corresponding UV-vis spectra (c, d) of pBTTT-C12 films, recorded for as-cast films, then heated from 25 °C to 175 °C in temperature steps of, respectively, 10 °C (WAXS) and 25 °C (UV-vis), and subsequent cooling to room temperature (b and d). | ||
Clearly, processing pBTTT-C12 in its mesophase results in considerable higher molecular order, thus raising the question what may be suitable annealing times to impart optimum bulk charge transport. We therefore performed TOF photoconductivity measurements on pBTTT-C12 structures that were annealed at 175 °C for various periods of time. From the resulting data, we conclude that rather extended annealing times are necessary for realizing both high molecular order and good overall charge transport in pBTTT-C12 bulk architectures (Fig. 3). Indeed, heat treatments of 2 h and less still resulted in essentially the same photo-electronic response as observed in as-cast pBTTT-C12. A strong qualitative improvement of charge transport in the form of reduced dispersion was observed only for architectures heat-treated for 7 h and more (Fig. 3b). Somewhat shorter TOF arrival times compared to as-cast structures—and as a consequence, higher room temperature hole mobilities—were also found for such long-time annealed pBTTT-C12 (Fig. 3b and c). Interestingly, the relative increase in mobility (about a factor of 5 to 10, depending on the field applied) for bulk, out-of-plane charge transport upon annealing is comparable to that observed in FET devices.6 Obviously, the absolute mobility values differ greatly (∼10−5 cm2 V−1 s−1 in TOF, compared to 0.2–0.6 cm2 V−1 s−1 in FETs), which we attribute to the highly anisotropic molecular architectures of the ordered pBTTT-lamella structures. Indeed, good charge transport can be expected in-plane of such pBTTT stacks but not out-of-plane, perpendicular to these supra-molecular arrangements.6
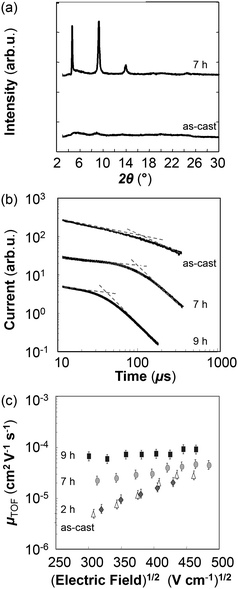 | ||
| Fig. 3 (a) Wide angle X-ray diffraction of pBTTT-C12 thin-film architectures. A comparison is shown between the as-cast polymer and pBTTT-C12 thermally treated for a period 7 hours. (b) Room temperature time-of-flight photocurrent transients for pBTTT-C12 structures, as-cast and after subjecting them to a range of annealing times (2, 7 and 9 h, film thickness of 3.9 to 5.1 µm, and electric field ≈ 1.6 × 105 V cm−1). (c) Corresponding Poole–Frenkel plots of room temperature hole mobility of such pBTTT-C12 architectures. | ||
The heat treatment in addition resulted in a distinctly different field and temperature behaviour (compare Fig. 1c and d). In fact, pBTTT-C12, annealed for 7 h at 175 °C, displayed a significantly smaller temperature dependence of µTOF in comparison with the as-cast architectures, apparently being less affected by the solid–solid phase transitions observed in pBTTT-C12. Moreover, a less pronounced field-dependence in mobility was found at room temperature for these heat-treated structures in accord with a higher ordered molecular arrangement.
Finally, in order to get additional information on the influence of annealing on pBTTT-C12, we also attempted to estimate the electronic disorder of such architectures by analysing the electric field and temperature dependence of the mobility using the correlated disorder model by Novikov and co-workers.18 To this end, some of the relevant transport parameters (energetic disorder, σ, positional disorder, 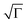 , prefactor mobility, µ0, and intersite distance, R) were extracted for both as-cast and annealed (7 h/175 °C) pBTTT-C12 films.19,20 As expected from the above discussion, the non-annealed pBTTT-C12 was found to be considerably disordered, both energetically (σ = 148 meV) and positionally (
, prefactor mobility, µ0, and intersite distance, R) were extracted for both as-cast and annealed (7 h/175 °C) pBTTT-C12 films.19,20 As expected from the above discussion, the non-annealed pBTTT-C12 was found to be considerably disordered, both energetically (σ = 148 meV) and positionally (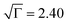 ), having a large intersite distance (R = 2.8 nm) supporting our WAXS and UV-vis spectroscopy data. Annealing (7 h/175 °C) resulted in significantly enhanced order (σ = 98 meV,
), having a large intersite distance (R = 2.8 nm) supporting our WAXS and UV-vis spectroscopy data. Annealing (7 h/175 °C) resulted in significantly enhanced order (σ = 98 meV, 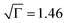 ) with a smaller intersite distance (R = 1.2 nm). The latter is consistent with the carriers being localised to a single monomer unit with an efficient overlap between units. The enhancement in positional order,
) with a smaller intersite distance (R = 1.2 nm). The latter is consistent with the carriers being localised to a single monomer unit with an efficient overlap between units. The enhancement in positional order, 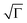 , may be explained by the observed microscopic changes occurring upon annealing, which DeLongchamp et al. attributed to an increase in the backbone order and π–π* stacking,12 however, we like to emphasis that they investigated thin films in the order of a few tens of nanometres, thus, microstructure formation might have been significantly influenced by the substrate, which is possibly less critical in thicker architectures as explored here. Finally, we attribute the improvement in energetic order, σ, deduced from our Novikov analysis to the reduction in the distribution of carrier delocalisation lengths (conjugation lengths) within sufficiently annealed pBTTT-C12 structures.16 We note, though, one internal inconsistency within the model, given the site distances obtained, namely the larger prefactor mobility calculated for the unannealed sample (µ0 = 0.088 cm2 V−1 s−1) compared to the annealed case (µ0 = 0.011 cm2 V−1 s−1). This might result from the fact that our transients are relatively dispersive, as discussed above, whilst the Novikov model was derived for non-dispersive charge transport, however, other factors or phenomena cannot be excluded.
, may be explained by the observed microscopic changes occurring upon annealing, which DeLongchamp et al. attributed to an increase in the backbone order and π–π* stacking,12 however, we like to emphasis that they investigated thin films in the order of a few tens of nanometres, thus, microstructure formation might have been significantly influenced by the substrate, which is possibly less critical in thicker architectures as explored here. Finally, we attribute the improvement in energetic order, σ, deduced from our Novikov analysis to the reduction in the distribution of carrier delocalisation lengths (conjugation lengths) within sufficiently annealed pBTTT-C12 structures.16 We note, though, one internal inconsistency within the model, given the site distances obtained, namely the larger prefactor mobility calculated for the unannealed sample (µ0 = 0.088 cm2 V−1 s−1) compared to the annealed case (µ0 = 0.011 cm2 V−1 s−1). This might result from the fact that our transients are relatively dispersive, as discussed above, whilst the Novikov model was derived for non-dispersive charge transport, however, other factors or phenomena cannot be excluded.
Summarising, our results illustrate that liquid crystallinity can be a powerful feature of polymer semiconductors to impart the ability to influence their molecular order and, thus, their electronic properties. Indeed, fastest bulk charge-transport in pBTTT-C12 was observed in its smectic-like12 phase above 150 °C. Therefore, high-mobility room-temperature structures may be designed by molecular tailoring derivatives of the interesting class of pBTTT polymers, such that the liquid-crystalline phases occur closer to the application/usage temperatures.
Nonetheless, even for systems such as the pBTTT-C12 with a mesophase at more elevated temperatures, liquid crystallinity can be exploited through suitable heat treatment procedures. To this end, we like to emphasize that in the case of pBTTT-C12 significantly longer annealing times were required to realize good bulk charge transport compared to the heat treatment procedures used for FET device fabrications. The duration of this heat treatment may be varied, e.g., by adjusting the molecular weight Mw of the polymer. However, we like to note that the pBTTT-C12 used was based on relatively short macromolecules (Mw ≈ 30 kg mol−1), which therefore most likely were unentangled. It is thus unclear if annealing times could be drastically reduced by lowering Mw. Nevertheless, our data strongly imply that in any case, a good compromise must be found between molecular order, optimization in charge transport, manufacturing throughput and, not to forget, material degradation due to long-time exposure to elevated temperatures.
Materials and Methods
Materials
Poly(2,5-bis(3-dodecyl-thiophen-2-yl)thieno[3,2-b]thiophene) (Mn = 27.9 kg mol−1, Mw = 51.4 kg mol−1) was generously supplied by Merck Chemicals. 1,2,4-Trichlorobenzene (TCB) was purchased from Aldrich and used as received.Sample preparation
Homogeneous thin films for TOF photoconduction experiments were prepared by first dissolving pBTTT-C12 (total polymer content: 0.5 wt%) in TCB at ∼80 °C. The hot solutions were then cast onto ITO-coated substrates kept at 50 °C until the solvent had evaporated. This resulted in films of 5–6 µm thick (measured with a Veeco Dektak3 ST surface profile measuring system). 50 nm thin counter-electrodes were then thermally evaporated. For wide-angle X-ray diffraction, films of approximately 0.5 to 2 µm were prepared accordingly, using same solution concentration. Finally, for temperature dependent UV-vis spectroscopy, films of a thickness of ∼100 nm were spin-coated from a 0.5 wt% hot (∼80 °C) solution first at 500 rpm for 30 seconds, followed by 2000 rpm for 30 seconds.Thermal analysis
Differential scanning calorimetry (DSC) was conducted on as-cast films (prepared as described above) under nitrogen at a scan rate of 10 °C min−1 with a Mettler Toledo DSC822 instrument. The sample weight was ∼5 mg.Wide-angle X-ray diffraction
Standard transmission wide-angle X-ray diffraction was carried out with an X'Pert Pro—PAN analytical instrument using CuKα-radiation (λ = 1.5418 Å), equipped with an Anton Parr HTK16 furnace operating between 25 °C and 175 °C at a rate of 10 °C per minute. Platinum heating strips were used as substrates with Pt/10% RhPt thermocouple welded to the underside of the strip to give accurate temperature measurement and control. An Anton Parr TCU 2000 temperature control unit provided accurate temperature control.UV-vis spectroscopy
A Perkin Elmer Lambda 900 was utilised equipped with a temperature-controlled demountable liquid flow cell (TFC-S25). We employed a heating/cooling rate of 25 °C min−1. The measuring cell was purged with N2 to prevent degradation of the polymer during heating.Time-of-flight photoconductivity
All measurements were carried out in a nitrogen atmosphere. The 6 ns, 337 nm wavelength, pulsed output of an EG101 Lambda Physik gas laser provided the optical excitation to create electron–hole pairs within a penetration depth of ∼100 nm of the top electrode in the case for neat pBTTT-C12, the 6 ns, 532 nm wavelength output of a frequency doubled insert model Nd:YAG laser provided the optical excitation, as the pBTTT-C12 displays an absorption peak close to this wavelength. All measurements were carried out under a DC bias from a low noise supply. The laser pulse intensity was kept sufficiently low. As a consequence, the photogenerated charge was less than 10% of the charge stored across the sample (CV, where C is the sample capacitance and V the applied potential), avoiding space charge effects that would result in a non-uniform electric field. The transient current was measured as a voltage drop across a range of load resistors (typically 47 Ω for fast signals and 2.31 kΩ for slow signals) at the input of a gain 11 amplifier whose output was connected to an Agilent Infiniium digitizing oscilloscope. Signal averaging (typically over 128 pulses) and background subtraction were performed in order to minimize both random and coherent radio frequency noise. Charge carrier mobility µTOF was calculated using the expression:| µTOF = d2/V × ttr(1.1) | (1) |
Acknowledgements
We are deeply indebted to Paul Smith for invaluable discussions about this project, and Oasis for providing the ideal environment for the latter. In addition, we would like to thank Merck Chemicals for so generously supplying materials. MB and SB are grateful for Merck CASE-studentships. Finally, NS, MH and IMC acknowledge support from EPSRC (EP/F056648/2).References
- M. Grell, W. Knoll, D. Lupo, A. Meisel, T. Miteva, D. Neher, H. G. Nothofer, U. Scherf and A. Yasuda, Adv. Mater., 1999, 11, 671 CrossRef CAS.
- R. J. Kline and M. D. McGehee, Polym. Rev., 2006, 46, 27 Search PubMed.
- H. Sirringhaus, R. J. Wilson, R. H. Friend, M. Inbasekaran, W. Wu, E. P. Woo, M. Grell and D. D. C. Bradley, Appl. Phys. Lett., 2000, 77, 406 CrossRef CAS.
- Z. J. Zheng, K. H. Yim, M. S. M. Saifullah, M. E. Welland, R. H. Friend, J. S. Kim and W. T. S. Huck, Nano Lett., 2007, 7, 987 CrossRef CAS.
- M. Grell, M. Redecker, K. S. Whitehead, D. D. C. Bradley, M. Inbasekaran, E. P. Woo and W. Wu, Liq. Cryst., 1999, 26, 1403 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, St. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328 CrossRef CAS.
- I. McCulloch, M. Heeney, M. L. Chabinyc, D. M. Delongchamp, R. J. Kline, M. Colle, W. Duffy, D. Fischer, D. Gundlach, B. Hamadani, R. Hamilton, L. Richter, A. Salleo, M. Shkunov, D. Sparrowe, S. Tierney and W. Zhang, Adv. Mater., 2009, 21, 1091 CrossRef CAS.
- B. H. Hamadani, D. J. Gundlach, I. McCulloch and M. Heeney, Appl. Phys. Lett., 2007, 91, 243512 CrossRef.
- T. Umeda, D. Kumaki and S. Tokito, J. Appl. Phys., 2009, 105, 024516 CrossRef.
- J. E. Parmer, A. C. Mayer, B. E. Hardin, S. R. Scully, M. D. McGehee, M. Heeney and I. McCulloch, Appl. Phys. Lett., 2008, 92, 113309 CrossRef.
- I. W. Hwang, J. Y. Kim, S. Cho, J. Yuen, N. Coates, K. Lee, M. Heeney, I. McCulloch, D. Moses and A. J. Heeger, J. Phys. Chem. C, 2008, 112, 7853 CrossRef CAS.
- D. M. DeLongchamp, R. J. Kline, Y. Jung, E. K. Lin, D. A. Fischer, D. J. Gundlach, S. K. Cotts, A. J. Moad, L. J. Richter, M. F. Toney, M. Heeney and I. McCulloch, Macromolecules, 2008, 41, 5709 CrossRef CAS.
- D. M. DeLongchamp, R. Joseph Kline, Y. Jung, D. S. Germack, E. K. Lin, A. J. Moad, L. J. Richter, M. F. Toney, M. Heeney and I. McCulloch, ACS Nano, 2009, 3, 780 CrossRef CAS.
- D. M. DeLongchamp, R. Joseph Kline, E. K. Lin, D. A. Fischer, L. J. Richter, L. A. Lucas, M. Heeney, I. McCulloch and J. E. Northrup, Adv. Mater., 2007, 19, 833 CrossRef CAS.
- M. L. Chabinyc, M. F. Toney, R. J. Kline, I. McCulloch and M. Heeney, J. Am. Chem. Soc., 2007, 129, 3226 CrossRef CAS.
- T. Kreouzis, D. Poplavskyy, S. M. Tuladhar, M. Campoy-Quiles, J. Nelson, A. J. Campbell and D. D. C. Bradley, Phys. Rev. B: Condens. Matter, 2006, 73, 235201 CrossRef.
- J. Clark, C. Silva, R. H. Friend and F. C. Spano, Phys. Rev. Lett., 2007, 98, 206406 CrossRef.
- S. V. Novikov, D. H. Dunlap, V. M. Kenkre, P. E. Parris and A. V. Vannikov, Phys. Rev. Lett., 1998, 81, 4472 CrossRef CAS.
- S. Barard, M. Heeney, L. Chen, M. Colle, M. Shkunov, I. McCulloch, N. Stingelin, M. Philips and T. Kreouzis, J. Appl. Phys., 2009, 105, 13701 CrossRef.
- R. U. A. Khan, D. Poplavskyy, T. Kreouzis and D. D. C. Bradley, Phys. Rev. B: Condens. Matter, 2007, 75, 35215 CrossRef.
- R. J. Baldwin, T. Kreouzis, M. Shkunov, M. Heeney, W. Zhang and I. McCulloch, J. Appl. Phys., 2007, 101, 23713 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |